基于多孔金结构的三相界面酶电极的制备及高效电化学酶传感性能
2021-10-15张嘉懿丁臻尧王丹丹陈礼平封心建
张嘉懿,丁臻尧,王丹丹,陈礼平,封心建,2
(1.苏州大学材料与化学化工学部,2.苏州大学化学科学国际合作创新中心,苏州215123)
开发高性能生物传感器对于精准医疗和人民健康具有重要意义[1~7].生物酶因具有专一性好和反应条件温和等优点而被广泛用于开发酶生物传感器[8~10].在天然氧气的存在下,氧化酶能选择性地氧化其底物(待测物),并生成等比例的过氧化氢,因此通过电化学方法测定酶催化反应产物过氧化氢的生成量即可获得待测物的浓度,这也是第一代酶传感器的基本工作原理[11~14].在酶传感检测中,酶催化反应通常发生在电极/溶液两相界面,界面反应所需的氧气由溶液提供.在氧化酶量一定的情况下,随着待测物浓度的增加,酶催化反应对氧气的需求量会逐渐上升,然而受限于溶液中氧气溶解度低和扩散速率慢等缺点,反应界面的氧气无法得到快速补给,严重限制了酶催化反应动力学以及产物过氧化氢的形成,进而限制了酶传感器检测的线性范围以及灵敏度.另外,由于常见待测溶液中氧气浓度易波动,严重影响了酶催化反应的稳定性,进而限制了酶传感器检测的准确性.
材料的表界面状态对包括酶催化反应在内的多相催化反应有重要的影响[15~17].自然界中许多生物体的表面(如荷叶)利用特定微纳米结构和低表面能物质的协同效应而呈现超疏水的性质[18~21].受该特性启发,研究者开发了多种具有超疏水性能的人工材料,并广泛用于自清洁材料[22]、电催化[23~25]以及光电催化[26,27]等物理、生物及化学研究领域.超疏水材料具较强的憎水/亲气性,当其浸入水溶液中时,会在液体和固体的界面处形成与大气连通的稳定气膜,即形成固-液-气三相共存的界面[28~33].相比于传统的固-液两相界面体系,这一独特的三相界面结构使得气体反应物能够通过气相快速传输至固-液反应界面[34~38],进而大幅提高气体参与的界面化学反应动力学.
本文通过电沉积法制备三维多孔金结构电极基底,对其表面进行超疏水和亲水润湿性调控,并负载氧化酶层,构建了具有固-液-气三相界面微环境的酶电极,实现了界面氧气的高浓度稳定输运及酶传感性能的大幅提高.该三相界面酶电极的工作原理如Scheme1所示,当该酶电极浸入待测溶液中时,会在超疏水多孔金基底内形成与大气连通的稳定气膜,电子受体氧气可以通过气相传输到酶催化反应界面.在氧气存在下,氧化酶会选择性地氧化其底物(待测物),同时生成相应比例的过氧化氢.由于氧气在空气中的含量远高于其在溶液中的溶解氧含量,并且氧气在气相中的扩散速率比其在液相中高104倍[39],这使得反应界面处的氧气浓度将从液相依赖(浓度低且易波动)变为气相依赖(浓度高且稳定).三相界面酶电极的构筑将极大提高氧化酶催化反应的动力学及其稳定性,进而可以大幅提高酶传感器检测的线性范围、灵敏度及准确性.
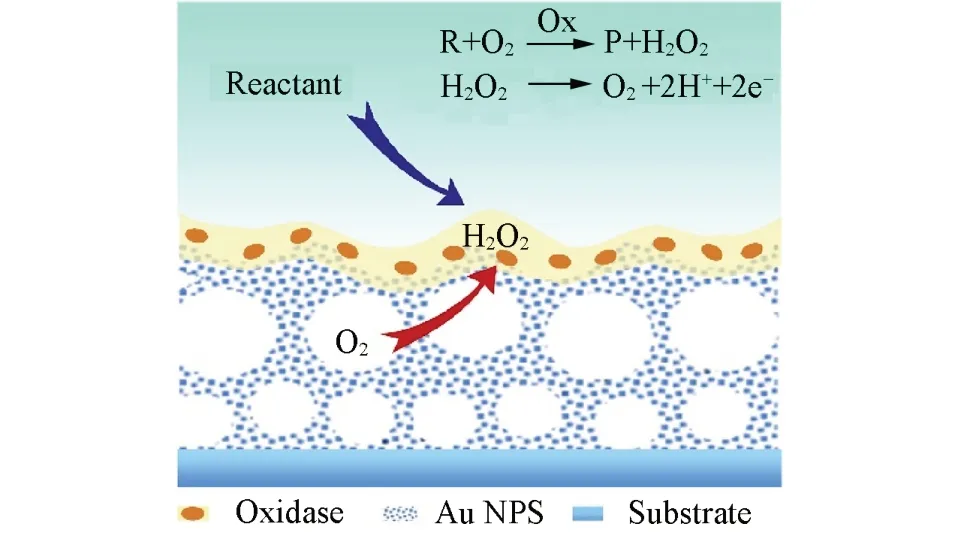
Scheme 1 Schematic illustration of the solid⁃liquid⁃air triphase enzyme electrode based on a three⁃dimensional(3D)porous Au substrate
1 实验部分
1.1 试剂与仪器
1H,1H,2H,2H-全氟辛基三氯硅烷(1H,1H,2H,2H-Perfluorooctyl trichlorosilane,97%,Macklin公司);三水合四氯金酸(HAuCl4·3H2O,98%,安耐吉化学公司);过氧化氢(H2O2,30%,永华化学股份有限公司);D-(+)葡萄糖(C6H12O6,99%)、氯化铵(NH4Cl,99.8%)、环己烷(C6H12,99.5%)、二水合磷酸二氢钠(NaH2PO4·2H2O,99%)、无水磷酸氢二钠(Na2HPO4,99%)和氯化钾(KCl,99.5%)(国药集团化学试剂有限公司);实验室用水为超纯水(电阻率≥18.2 MΩ·cm,利用上海和泰仪器有限公司Master-S15纯水仪制备).
CHI 660E型电化学工作站(上海辰华仪器有限公司);GVC-2000型离子溅射仪(上海禾早电子科技有限公司);Evolution220型紫外-可见分光光度计(美国Thermo Fisher公司);SU8010型扫描电子显微镜(日本Hitachi公司).
1.2 多孔金结构三相界面酶电极的制备
1.2.1 多孔金电极基底的制备参照文献[40]方法,在10 mmol/L HAuCl4与2.5 mol/L NH4Cl的混合溶液中,以金片为工作电极,铂丝为对电极,Ag/AgCl(3 mol/L KCl)为参比电极,采用循环伏安法,以50 mV/s的扫描速率在+0.8~0 V之间进行25次扫描.然后在−2.5 V电位下继续沉积一定时间(0~120 s).
1.2.2 多孔金基底的超疏水处理在10 mL环己烷中加入20 μL 1H,1H,2H,2H-全氟辛基三氯硅烷,将上述多孔金基底在该混合液中浸泡2 h,取出后放入120℃烘箱中处理2 h,得到具有超疏水性的多孔金基底.
1.2.3 过氧化氢电催化剂的沉积使用离子溅射仪在超疏水多孔金表面溅射铂(Pt)纳米颗粒,工作电流为20 mA,溅射时间为45 s.
1.2.4 葡萄糖氧化酶的负载取体积为100 μL的葡萄糖氧化酶(GOx)溶液(20 mg/mL),加入50 μL壳聚糖溶液(2 mg/mL,1%的乙酸溶液),再加入45 μL去离子水和5 μL戊二醛溶液(质量分数5%),配制葡萄糖氧化酶-壳聚糖(GOx-Chit)混合液.取不同量的GOx-Chit混合液(体积分别为1,3,5,7,10 μL)滴在沉积了Pt催化剂的多孔金基底表面,室温下干燥,即获得三相界面葡萄糖氧化酶电极.
两相界面酶电极采用了类似的构筑方法,只是没有对多孔金基底进行超疏水处理.
1.3 过氧化氢生成速率的测定
在磷酸盐缓冲溶液(PBS,0.2 mol/L Na2HPO4-NaH2PO4)中加入不同浓度的葡萄糖,将三相界面酶电极浸入溶液,以600 r/min的速度搅拌,10 min后取840 μL反应液加入比色皿,依次加入375 μL邻苯二甲酸氢钾和375 μL碘化钾/氢氧化钠/钼酸钠混合液(1.2 mol/L碘化钾溶液、0.18 mol/L氢氧化钠溶液和0.3 mmol/L七钼酸钠溶液按体积比1∶1∶1混合).再以500 r/min的速度搅拌5 min后,采用紫外-可见分光光度计测定混合液的吸光度,并通过标准曲线对过氧化氢的产量进行标定.
1.4 电化学检测
在PBS溶液中,以三相界面酶电极为工作电极,铂丝为对电极,Ag/AgCl(3 mol/L KCl)电极为参比电极,加入不同浓度的待测液后,搅拌30 s,静置10 s后,检测相应电位下的电流值.
2 结果与讨论
2.1 多孔金基底及三相界面酶电极的结构表征
图1 (A)和(B)分别为电沉积法制备的多孔金结构的低倍和高倍扫描电子显微镜(SEM)照片.从图中可以看到金膜基底呈三维多孔状结构,孔洞尺寸约为48~56 μm(图S1,见本文支持信息),多孔结构是由平均粒径为43.6 nm的金颗粒组装而成,多孔膜的厚度约为28.2 μm,且各层孔洞间相互连通(图S2,见本文支持信息).这是由于在电沉积过程中,H+和金离子[Au(III)]在阴极同时被还原,并产生大量的氢气.氢气气泡在脱离基底过程中充当Au(III)还原的动态模板,从而形成多孔网状结构.

Fig.1 SEM images of porous Au substrate and triphase enzyme electrode
图1 (A)插图为水滴经疏水处理后多孔金表面的光学照片.水在其表面的接触角为(151±2)°,表明经过1H,1H,2H,2H-全氟辛基三氯硅烷修饰后的多孔金表面具有超疏水特性.在超疏水多孔金表面进一步溅射Pt纳米颗粒(过氧化氢电催化剂,图S3,见本文支持信息),并通过X射线光电子能谱进行元素分析(图S4,见本文支持信息).图S4(A)为所制电极的元素全谱扫描图,从图中可以看出金(Au)和铂(Pt)元素均存在于电极中.图S4(B)和(C)分别为Au4f和Pt4f的XPS谱[41].在溅射Pt纳米颗粒的电极表面负载葡萄糖氧化酶,获得了三相界面酶电极.图1(C)和(D)分别为多孔金结构三相界面酶电极的表面和侧面SEM照片,从图中可见酶层均匀铺展在多孔金表面,且酶电极表面呈现亲水性,水在酶电极表面的接触角为(35±2)°[图1(C)插图].从图1(D)中可以看出,多孔金表面覆盖有酶层,但是底部仍然保持多孔形貌.为了更直观地表征三相界面,将三相界面酶电极浸泡在罗丹明B(RhB)溶液中,使亲水的氧化酶层染色,并通过激光共聚焦显微镜采用层扫模式从上到下间隔1 μm进行连续扫描,收集酶层吸附的RhB染料分子的荧光信号(图S5,见本文支持信息).激光共聚焦显微镜由图S5(A)中的扫描方向自上而下进行层扫,得到的扫描图片如图S5(B)所示.其中,酶层中心区域[图S5(A)中每个小图的中间区域]最初荧光信号较弱(Layer 1),随后扫描信号开始逐渐增强(Layers 2和3),最后所选区域中心的荧光信号完全消失(Layer 4).该结果表明,吸附了RhB分子的酶层只能浸润电极表面,无法浸润电极内部,证明只有电极表面的区域亲水,而内部保持疏水.
因此,当将该酶电极没入待测溶液中时,溶液可以浸润顶部的亲水酶层,无法浸润下部疏水的多孔金(图S6,见本文支持信息),这一独特的三相界面酶电极结构使得氧气可以直接从大气环境通过气相快速传输到氧化酶催化反应界面,提高酶催化反应速率和稳定性,进而提高酶传感检测的性能.在传统固-液两相界面酶电极的传感体系中(图S7,见本文支持信息),氧化酶催化反应所需要的氧气只能通过液相缓慢且不稳定地供给.
2.2 三相界面氧化酶催化性能探究
通过测量酶催化反应产物过氧化氢的生成速率,考察了基于三相界面的酶催化活性.通过碘化法对酶催化反应产物过氧化氢进行显色,并利用紫外-可见吸收光谱测量显色产物的吸光度变化,可以得到酶催化反应中过氧化氢的生成速率.如图2(A)所示,在三相界面酶催化反应体系中,随着葡萄糖浓度的增加,350 nm处的特征吸收峰位置保持不变,但吸光度逐渐增加,表明过氧化氢的浓度在逐渐增加.通过Lambert-Beer定律测量得到酶电极在特定葡萄糖浓度下一定时间内生成的过氧化氢量,并计算得到过氧化氢的反应速率v.图2(B)中红色曲线为三相界面复合电极在0~50 mmol/L葡萄糖浓度范围内所测得的过氧化氢生成速率,黑色曲线为两相界面复合电极在0~20 mmol/L葡萄糖浓度范围内所测得的过氧化氢生成速率,曲线拟合结果表明三相和两相界面酶电极的酶催化反应均遵循米氏(Michaelis-Menten)动力学模型.基于图2(B)中过氧化氢生成速率随葡萄糖浓度的变化曲线,根据米氏方程

的双倒数变形式

以1/v作对1/[S]作图,所得方程的截距即为该催化反应的最大反应速率vmax,继而根据下式

可求得催化反应速率常数kcat.其中,KM(mol/L),[S](mol/L),v(mol⋅L−1⋅min−1),[E](mol⋅L−1⋅min−1)分别为米氏常数、底物浓度、反应速率和酶浓度.由图2(B)中插图可知,两相界面酶电极测得的动力学参数为kcat=22.2 min−1,三相界面酶电极测得的动力学参数为kcat=600.9 min−1,即三相界面酶催化反应速率常数比两相界面提高了27倍.这主要是由于三相界面电极在浸入待测溶液中时,疏水的多孔金能存储氧气,使得氧气可以直接从气相传输至酶催化反应界面,酶催化反应所需氧气能得到迅速补充,进而大幅度提高了其反应动力学.
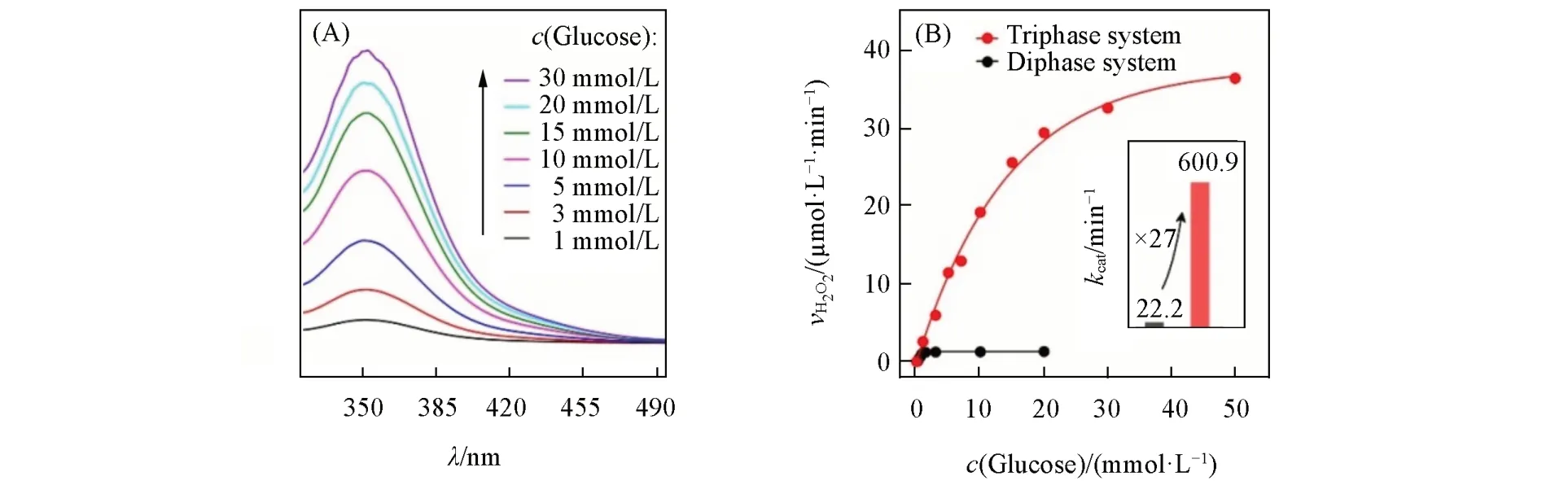
Fig.2 Oxidase kinetics of triphase and diphase nanoporous Au⁃based enzyme electrodes
2.3 多孔金结构三相界面酶电极的优化
进一步研究了不同沉积时间所制备的多孔金基底对酶催化反应的影响,并对三相界面酶电极结构进行了优化.图3(A)~(F)为沉积不同时间的多孔金基底的SEM照片,当沉积时间小于30 s时,金颗粒尚未组装形成三维多孔结构,厚度为8.3 μm(图S8,见本文支持信息),表面疏水处理后无法对气体的传输起到明显促进作用,因此无法有效提升酶催化反应速率[图3(G)].当电沉积时间增加到120 s时,以电化学还原持续产生的氢气气泡作为Au(III)还原的模板,可以形成完整的三维孔道结构.气体能通过疏水处理后的孔道快速传输至酶催化反应区域,最终提高酶催化反应速率.而当沉积时间继续增加至150 s时,虽然所沉积样品仍然具有三维多孔结构,但由此制备的三相界面酶催化反应速率并没有进一步增加.这可能是由于气体传输的瓶颈问题已被克服,更厚的多孔膜不会再带来额外的性能提升.另外还发现在电沉积过程中一旦沉积时间超过120 s,三维多孔金膜容易从导电基底脱落,不利于后续酶电极的稳定构筑.如图3(G)所示,随着电沉积多孔金的时间增加,复合电极酶催化反应产物过氧化氢在1~50 mmol/L葡萄糖中的生成速率逐渐增大,且电沉积时间为120 s时生成速率达到最大值;继续增加电沉积时间,过氧化氢的生成速率不再增大.通过电化学法测得不同沉积时间的多孔金基底对葡萄糖检测的影响,如图S9所示(见本文支持信息),随着电沉积多孔金的时间增加,响应电流逐渐增大,且电沉积时间为120 s时检测电流达到最大值;继续增加电沉积时间,葡萄糖检测电流不再增大,与酶动力学测试结果一致.因此选择沉积时间120 s作为多孔金电极的最佳电沉积时间.

Fig.3 SEM images of porous Au substrate electrodeposited at different times(A—F)and oxidase kinetics of triphase enzyme electrode based on these porous substrates(G)
在电沉积时间为120 s的多孔金基底上对GOx-chit的用量进行了优化.如图S10(见本文支持信息)所示,当GOx-chit的用量由1 μL增加到7 μL时,测定的酶催化反应产物过氧化氢的生成速率在1~50 mmol/L葡萄糖浓度下逐渐增加,且当Gox-chit的用量为7 μL时,过氧化氢生成速率达到最大值;继续增加GOx-chit用量至10 μL时,过氧化氢生成速率不再改变.因此,后续实验将控制GOx-chit用量为7 μL作为酶电极的最佳制备条件.
2.4 三相界面酶电极的传感检测性能
以葡萄糖为模型待测物,以三相界面酶电极为工作电极研究了其对葡萄糖的检测性能.图4(A)为三相界面酶电极对10 mmol/L葡萄糖的线性扫描伏安法(LSV)检测曲线.图中氧化电流随着检测电位的增加而增加,并在+0.4 V下电流开始出现平台,继续增大电位其响应电流不再明显增加,因此选取+0.4 V为后续葡萄糖检测的工作电位.如图4(B)所示,在检测电位为+0.4 V时,随着葡萄糖浓度的增加,氧化电流逐渐增大,选择第8 s读取的电流值对底物浓度作图,得到图4(C)中的红色曲线.可见,三相界面酶传感体系的线性检测范围可以达到80 mmol/L(R2=0.99),灵敏度为9.43 μA·mmol−1·L−1,表明该体系对葡萄糖检测具有良好的线性关系和较宽的线性范围.
图4 (C)插图为两相界面酶电极体系检测葡萄糖的电流-浓度曲线.可以看到其线性检测范围仅为1.6 mmol/L,远低于三相界面传感体系.这是由于在传统固-液两相界面反应体系中,氧气在溶液中含量低且传输速率慢,在高浓度底物的测试环境下氧化酶反应所需的氧气无法得到及时快速补给,严重抑制了氧化酶的催化活性.相比之下,对于固-液-气三相界面酶催化反应体系,氧气能通过气相快速供给到反应界面,满足高浓度葡萄糖条件下酶催化反应的需要,并产生大量的过氧化氢.因此,基于多孔金结构三相界面酶电极的传感体系表现出了较宽的检测线性范围和高的灵敏度.
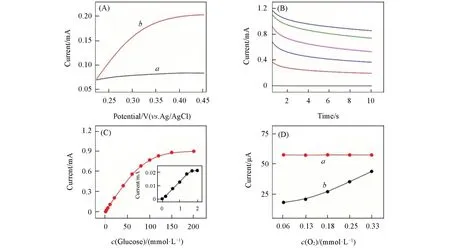
Fig.4 Detection performance of triphase nanoporous Au biosensor
溶液中氧气浓度的波动对酶催化反应的稳定性以及酶电极的检测稳定性有很大影响.接下来通过在溶液中通入氮气调节溶解氧浓度,考察了酶电极的抗溶解氧浓度波动的性能.如图4(D)中黑色曲线所示,两相界面酶电极体系的检测电流随着氧浓度的升高而增大,表明氧气浓度波动对传统固-液两相界面酶催化反应速率及酶电极响应性能有很大的影响,限制了酶电极检测的稳定性.而同样如图4(D)所示,三相界面酶电极在不同溶解氧浓度情况下,对同一浓度葡萄糖的氧化电流响应基本保持稳定(红色曲线),表明三相反应界面体系中所需氧气可以稳定地从气相中得到补充,摆脱了液相溶解氧浓度波动而造成的不利影响,从而具有高的检测稳定性.
最低检出限是酶电极传感性能的一项重要指标,在检测电位为+0.4 V时,通过i⁃t法测得本文构建的三相界面酶传感体系对葡萄糖的检出限为2 μmol/L[图S11(A),见本文支持信息],这表明多孔金结构三相酶电极不仅可以对高浓度待测物进行检测,还可以对低浓度待测物进行检测.重现性也是酶传感检测的一项重要指标.对同一个三相界面酶电极在含5 mmol/L葡萄糖的PBS溶液中连续进行150次测量,考察了其重现性,结果表明,150次检测电流的相对标准偏差仅为1.0%[图S11(B)],表明该三相界面酶电极体系具有良好的重现性.
通过与已报道的用于葡萄糖检测的酶电极传感性能进行比较(表S1,见本文支持信息)可以看出,传统固-液两相界面的酶电极中,由于反应物氧气供给不足,传感器的检测上限较低.本研究引入固-液-气三相界面,解决了氧气供给的问题,提升了传感器检测的线性范围,同时也保证了较高的灵敏度,实现了高灵敏度和宽线性范围的葡萄糖检测.
3 结 论
采用电沉积法制备了具有三维多孔结构的导电金基底,通过对其表面进行超疏水和亲水润湿性调控并负载氧化酶层,构建了具有固-液-气三相界面微环境的酶电极.三相界面能够为酶催化反应所需要的氧气提供快速的气相供给通道,使得反应界面氧气浓度从液相依赖(浓度低且波动)变为气相依赖(浓度高且稳定),显著提高了酶催化反应的速率和稳定性.基于该三相界面酶电极实现了对葡萄糖的宽线性范围、高灵敏度和高准确性的检测.本研究表明反应界面微环境的设计和调控对提升酶催化反应性能以及开发高效酶传感器具有重要意义.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/20210355.
