碳掺杂六方氮化硼/二硫化钼吸附还原六价铬和助催化降解有机污染物
2021-10-15崔金萍陈温贤郁非繁曹诗雨吕维扬姚玉元
崔金萍,陈温贤,郁非繁,曹诗雨,吕维扬,姚玉元
(浙江理工大学纺织纤维材料与加工技术国家地方联合工程实验室,杭州310018)
随着经济的快速发展,电镀、皮革、冶金和纺织印染等行业产生了大量含铬废水[1,2].废水中铬主要以Cr(Ⅲ)与Cr(Ⅵ)的形式存在,其中Cr(Ⅲ)毒性较低[3],是哺乳动物不可或缺的一种微量营养元素,而Cr(Ⅵ)具有高毒性、致癌致畸和不可生物降解的特性,严重威胁到环境安全和人类健康[4],所以必须对废水中的Cr(Ⅵ)进行严格处理后才能排放.目前,处理废水中Cr(Ⅵ)的主要方法有吸附法、膜过滤法和吸附还原法[5,6],其中吸附还原法是一种将吸附的Cr(Ⅵ)转变为Cr(Ⅲ)以达到降低Cr(Ⅵ)生态毒性的有效方法[7,8].因此,吸附还原法成为Cr(Ⅵ)处理领域的研究热点和重要发展方向.
二硫化钼(MoS2)是一种类石墨烯二维过渡金属硫化物[9],具有独特三层堆叠原子层(S-Mo-S)的夹层结构及优异的化学稳定性和结构可设计性,而且MoS2表面和边缘丰富的硫基团赋予其良好的吸附性能[10,11],因此,近年来MoS2成为吸附还原去除废水中Cr(Ⅵ)的理想材料.Wang等[12]采用水热法制备了Mg/Al层状双金属氢氧化物改性的二硫化钼复合材料,去除水溶液中Cr(Ⅵ)的最大容量为76.30 mg/g.Li等[13]制备聚吡咯包覆二硫化钼,去除Cr(Ⅵ)的最大容量为257.73 mg/g.Wang等[14]研究了聚乙烯吡咯烷酮插层MoS2复合材料和聚丙烯酰胺插层MoS2复合材料去除水溶液中的Cr(Ⅵ),去除最大容量分别为142.24和84.91 mg/g.但是,这些复合改性纳米MoS2处理Cr(Ⅵ)还存在去除容量低、去除速率慢等缺点.因此,如何提高MoS2处理Cr(Ⅵ)的去除容量和去除速率仍然是重金属废水污染领域极具挑战性的研究课题.
六方氮化硼(BN)是一种类石墨结构的层状材料,具备良好的吸附、导热、高机械强度及化学惰性等特点,而且在高温和强光照条件下还具有高抗氧化性[15,16],上述特征使其可能成为一种优良的催化剂载体,但由于BN表面比较光滑,导致其表面发生化学反应的能力较低.多巴胺是一种性能优异的表面改性剂,在空气中容易发生自聚反应,在基体表面包覆一层富含碳氮元素的聚多巴胺(PDA)膜.本文以六方氮化硼为载体,采用PDA对六方氮化硼改性并经煅烧制得碳掺杂六方氮化硼(c-BN),结合水热法得到碳掺杂六方氮化硼负载MoS2的纳米复合材料(c-BN@MoS2),其制备路线图见Scheme 1.研究发现,c-BN@MoS2能够快速去除废水中的Cr(Ⅵ),而且去除容量明显高于多数已报道的MoS2复合材料;c-BN@MoS2还可大幅提高Fe2+/PMS类芬顿的催化性能.所制备的c-BN@MoS2呈现出优异的吸附还原六价铬和高效降解抗生素等污染物的双功能特性,拓展了六方氮化硼在废水处理中应用的新方向.
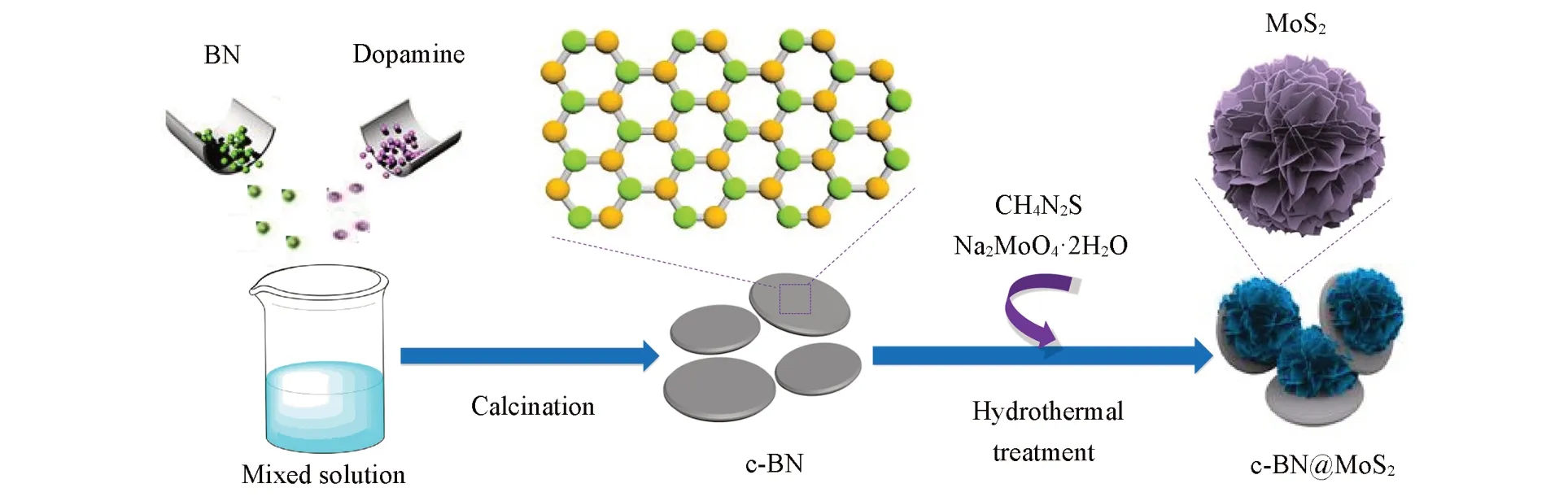
Scheme 1 Schematic preparation process of c⁃BN@MoS2
1 实验部分
1.1 试剂与仪器
六方氮化硼(BN)、钼酸钠二水化合物(Na2MoO4⋅2H2O)、硫脲、二苯氨基脲、盐酸多巴胺、5,5-二甲基吡咯啉-氧化物(DMPO)、过一硫酸氢盐(PMS)、三(羟甲基)氨基甲烷和磺胺甲恶唑(SMX)均为分析纯,购于上海阿拉丁有限公司;重铬酸钾(K2Cr2O7)为分析纯,购于中国医药上海化学试剂公司.
1800型可见分光光度计(UV-1800),美国尤尼柯上海有限公司;UPLC型高效液相色谱仪,美国Waters公司;K-Alpha型X射线光电子能谱仪(XPS),美国Thermo Fisher Scientific公司;A300型电子顺磁共振波谱仪(EPR),德国Bruker公司;Optima 7300DV型电感耦合等离子体发射光谱仪(ICP),美国PerkinElmer公司;D8 Advance型X射线衍射仪(XRD),德国布鲁克AXS公司;Renishaw inVia型拉曼光谱仪(Raman),英国雷尼绍公司;GeminiSEM 300型场发射扫描电子显微镜(SEM),德国蔡司公司.
1.2 实验过程
1.2.1 c-BN的制备向0.01 mol/L、pH值为8.5的三(羟甲基)氨基甲烷缓冲溶液中加入0.5 g BN,超声处理30 min后,加入0.2 g盐酸多巴胺并振荡反应6 h后,经离心收集黑色沉淀物,在60℃干燥箱中干燥12 h,再将干燥后的产物放置于管式炉中,氩气保护下以5℃/min的升温速率加热至900℃并保温2 h,自然冷却至室温后即得样品c-BN.
1.2.2 c-BN@MoS2的制备称取0.2903 g Na2MoO4⋅2H2O和0.3425 g硫脲并溶解在38 mL去离子水中,将0.1 g c-BN加入到上述溶液中,经超声和搅拌后,将其转移至100 mL内衬为特氟龙的高压反应釜中,在200℃条件下反应22 h,冷却至室温,采用无水乙醇和去离子水多次洗涤去除表面残留物,然后在60℃干燥箱中干燥12 h,自然冷却至室温后即得样品c-BN@MoS2.
1.2.3 去除Cr(Ⅵ)的性能测试Cr(Ⅵ)溶液由K2Cr2O7配制而成,用硫酸和氢氧化钠调节pH值.量取40 mL Cr(Ⅵ)溶液,加入0.02 g c-BN@MoS2,在25℃,130 r/min的条件下进行去除反应.间隔一定时间后,用1 mL的注射器取样,通过显色法测定Cr(Ⅵ)浓度,采取电感耦合等离子体光谱仪测定溶液中Cr的总浓度.
1.2.4 催化性能测试配制20 μmol/L的SMX溶液,并量取20 mL置于玻璃瓶中,依次加入5 mg c-BN@MoS2,0.04 mmol/L Fe2+以及0.5 mmol/L PMS,在25℃,130 r/min的条件下进行降解反应,每间隔2 min取一次样,并注入2 mL液相瓶中,进行高效液相色谱分析.
2 结果与讨论
2.1 样品的结构表征
BN,c-BN,MoS2和c-BN@MoS2的SEM照片如图1所示.图1(A)为单独BN的SEM照片,图1(B)为BN经PDA改性并煅烧制得的c-BN的SEM照片,通过比较BN和c-BN的SEM照片可知,c-BN表面存在一层覆盖物,且表面变得比较粗糙,c-BN的TEM照片[见本文支持信息图S1(A)]进一步证实纳米片状的BN表面有覆盖物存在,推测PDA经煅烧后在BN表面沉积了一层碳氮材料.从MoS2的SEM照片[图1(C)]中可知,其呈现出花状的纳米球结构[17],并且紧密堆积在一起,在c-BN@MoS2的SEM照片[图1(D)]中,纳米球结构的MoS2分散开来,形成较小的花状结构,结合c-BN@MoS2的TEM照片[图S1(B)]可知,花状的MoS2生长在c-BN载体上.图1(E)为c-BN@MoS2的SEM照片,结合SEM元素分布图[图1(F)~(J)]可以看出,C,N,B,S和Mo元素分布比较均匀.

Fig.1 SEM images of different samples(A—D)and c⁃BN@MoS2(E),corresponding SEM elemental mappings of C(F),N(G),B(H),S(I),Mo(J)
采用XRD对样品的晶体结构、组成进行测试,结果如图2(A)所示.可见,制备的MoS2在2θ为14.60°,33.50°和58.92°处有特征峰,与MoS2标准卡片(JCPDS No.78-2177)对比,表明是MoS2的典型特征峰,可分别属于MoS2的(002),(100)和(110)晶面[12].与MoS2相比,c-BN@MoS2的(002),(100)和(110)衍射峰均向左发生了偏移,而且衍射峰降低,这可能是由于出现了1T相MoS2[18].此外,c-BN@MoS2在2θ为26.75°,43.85°处出现了明显的特征峰,这2个特征峰与单独c-BN的特征峰一致,其中在2θ=26.75°处的特征峰是由BN的(002)晶面和非晶碳[19]的(002)晶面共同衍射引起的,2θ=43.85°处的特征峰可属于BN的(100)晶面[20].XRD结果表明,样品具有较好的结晶性,且c-BN@MoS2仍保留了c-BN和MoS2的结构.
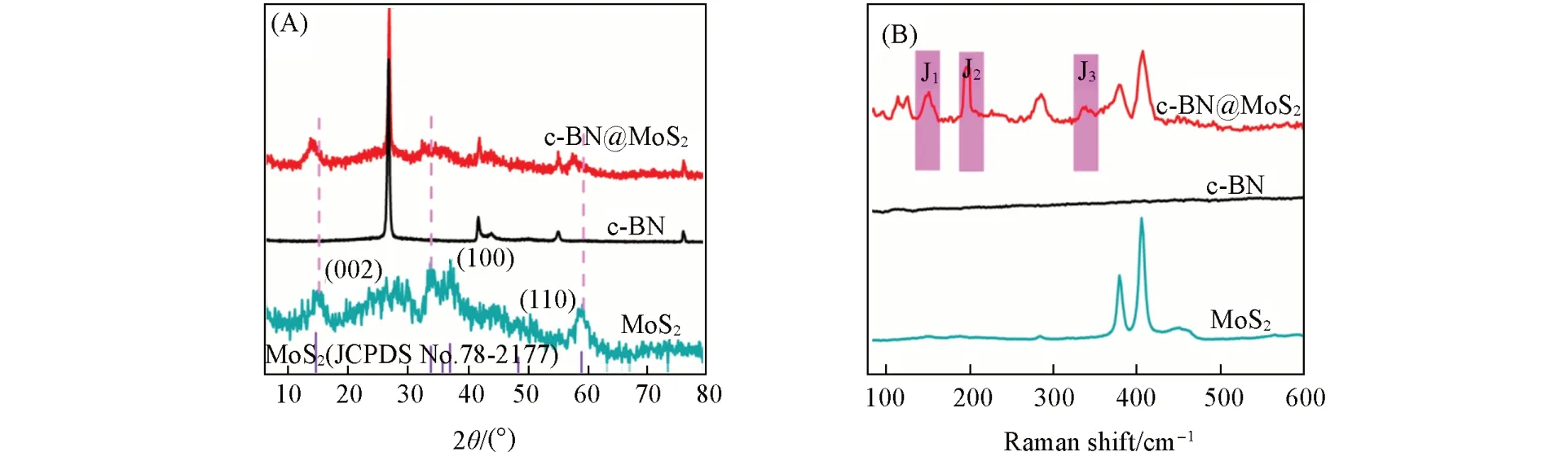
Fig.2 XRD patterns(A)and Raman spectra(B)of MoS2,c⁃BN and c⁃BN@MoS2
为了进一步分析c-BN@MoS2中MoS2的相转变,对MoS2,c-BN和c-BN@MoS2进行了拉曼光谱的测试,结果见图2(B).可见,单独MoS2在380和404 cm−1处有2个特征峰,可分别属于2H相MoS2的峰和峰[18].与MoS2和c-BN相比,c-BN@MoS2的峰明显减弱,同时在150,198,280,330 cm−1处出现了新的特征峰,分别对应于J1,J2,E1g和J3峰[21,22].此外,在图S2(见本文支持信息)中,c-BN@MoS2在1363,1597 cm−1处出现的2个特征峰,与c-BN的峰值一致,属于BN的E2g峰和非晶碳的G峰.上述分析进一步表明,c-BN@MoS2中出现了1T相MoS2.
利用XPS检测了c-BN@MoS2,MoS2的价态和组成.图S3(A)(见本文支持信息)是c-BN@MoS2的XPS全谱图,在谱图中含有C,N,O,Mo,S和B对应的特征峰,这与预期结果一致.图3(A)为N1s窄谱图,可见,在397.63,397.04和398.91 eV处出现特征峰,可分别对应于N—B键、吡啶N[23]和N—C键.在B1s的窄谱图[图S3(B)]中,呈现了处于190.07和189.53 eV处的特征峰,可归属于B—N键和B—C键.上述结果表明c-BN@MoS2中存在BN,且被PDA包裹的BN通过煅烧产生了吡啶N和碳.图3(B)为Mo的窄谱图,由图可见,c-BN@MoS2和MoS2中都有Mo3d3/2和Mo3d5/2的结合能峰,表明样品中存在Mo4+,在225.51 eV处有一个S2s的次要峰,结合S2p中[图3(C)]的特征峰,说明S2−的存在,因此可推测成功合成了MoS2.图3(D)为C1s的窄谱图,在283.46和286.36 eV处出现的特征峰可对应于C—B键和C—N键,进一步证实碳掺杂BN的存在.在285.12 eV处出现了属于C—O—Mo键的特征峰,表明MoS2与c-BN上的碳发生了反应.此外,单独MoS2在232.36和229.02 eV处的2个特征峰,可分别归属于2H相MoS2的Mo3d3/2和Mo3d5/2的结合能峰,而c-BN@MoS2在2H相MoS2的峰值明显降低,且在231.32和228.12 eV处出现了新特征峰,可对应于1T相MoS2的Mo3d3/2和Mo3d5/2特征峰[18].在S2p的窄谱图中,相比于MoS2,c-BN@MoS2在162.38和161.10 eV处也出现了1T相MoS2的的两个结合能峰.上述分析表明MoS2已成功负载到c-BN上,并证实c-BN@MoS2中MoS2以呈现金属特性的亚稳定1T相为主,明显不同于单独MoS2以2H相为主的稳定半导体结构特性,这有助于MoS2还原反应过程中的电子转移,为进一步高效去除废水中六价铬和降解有机污染物提供了可能.
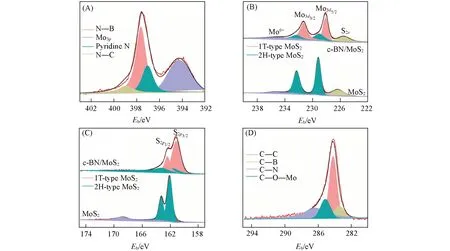
Fig.3 XPS spectra of c⁃BN@MoS2
2.2 c-BN@MoS2去除Cr(Ⅵ)的性能
为了考察c-BN@MoS2对Cr(Ⅵ)的吸附还原性能,将c-BN@MoS2加入到50 mg/L的K2Cr2O7溶液中,其去除效果如图4(A)所示.可见,c-BN@MoS2与Cr(Ⅵ)反应40 min时,可去除95%以上的Cr(Ⅵ),远高于BN(去除率11%),c-BN(去除率22%)和MoS2(去除率39%).图4(B)为BN,MoS2,c-BN和c-BN@MoS2对不同初始浓度Cr(Ⅵ)的去除容量(qt),与BN(去除容量23 mg/g),c-BN(去除容量39 mg/g)和MoS2(去除容量98 mg/g)相比,c-BN@MoS2去除Cr(Ⅵ)的容量达到401 mg/g,而且该去除容量明显优于已知的其它材料(见本文支持信息表S1).由此可见,MoS2负载到c-BN上,其去除速率和去除容量均远高于单独的MoS2,这说明c-BN载体对吸附还原Cr(Ⅵ)的作用十分重要.为了研究c-BN增强MoS2去除Cr(Ⅵ)的作用,对MoS2和c-BN@MoS2进行BET测试,结果如图S4(见本文支持信息)所示.由图可知,c-BN@MoS2平均孔径和比表面积分别为28.40 nm和21.53 m2/g,高于MoS2的平均孔径(11.45 nm)和比表面积(16.12 m2/g),因此载体c-BN可以提高MoS2的分散性,从而增多了还原活性位点数量.同时拉曼光谱和XPS分析显示,MoS2负载到c-BN上后生成了1T相为主的MoS2,这为加快还原过程中电子的转移提供了可能.
样品去除Cr(Ⅵ)之后的再生性能也是评价该样品的重要标准[24],为了考察c-BN@MoS2的再生性能,c-BN@MoS2在吸附还原Cr(Ⅵ)后,经离心、碱洗酸洗,再经水洗、烘干c-BN@MoS2后重新吸附还原Cr(Ⅵ),按照上述同样步骤重复4次,c-BN@MoS2去除Cr(Ⅵ)的性能示于图4(C).由图可知,再生循环5次后,c-BN@MoS2仍能去除84%的Cr(Ⅵ),表明c-BN@MoS2具有优异的重复再生性能.此外,研究了c-BN@MoS2在不同pH、离子、温度下去除Cr(Ⅵ)的性能,图4(D)表明,随着Cr(Ⅵ)溶液pH值的增加,c-BN@MoS2吸附还原Cr(Ⅵ)的容量明显降低,在pH为4时,去除容量大幅下降至65 mg/g.这是由于在不同pH值下c-BN@MoS2的Zeta电位不同所致[图4(D)],当pH值小于4时,c-BN@MoS2表面带正电,有助于吸附并还原Cr(Ⅵ)[25],从而提高其去除容量;当pH大于4,结果反之.温度和离子对Cr(Ⅵ)的去除性能几乎无影响(见本文支持信息图S5),表明c-BN@MoS2具有良好的环境抗干扰性.
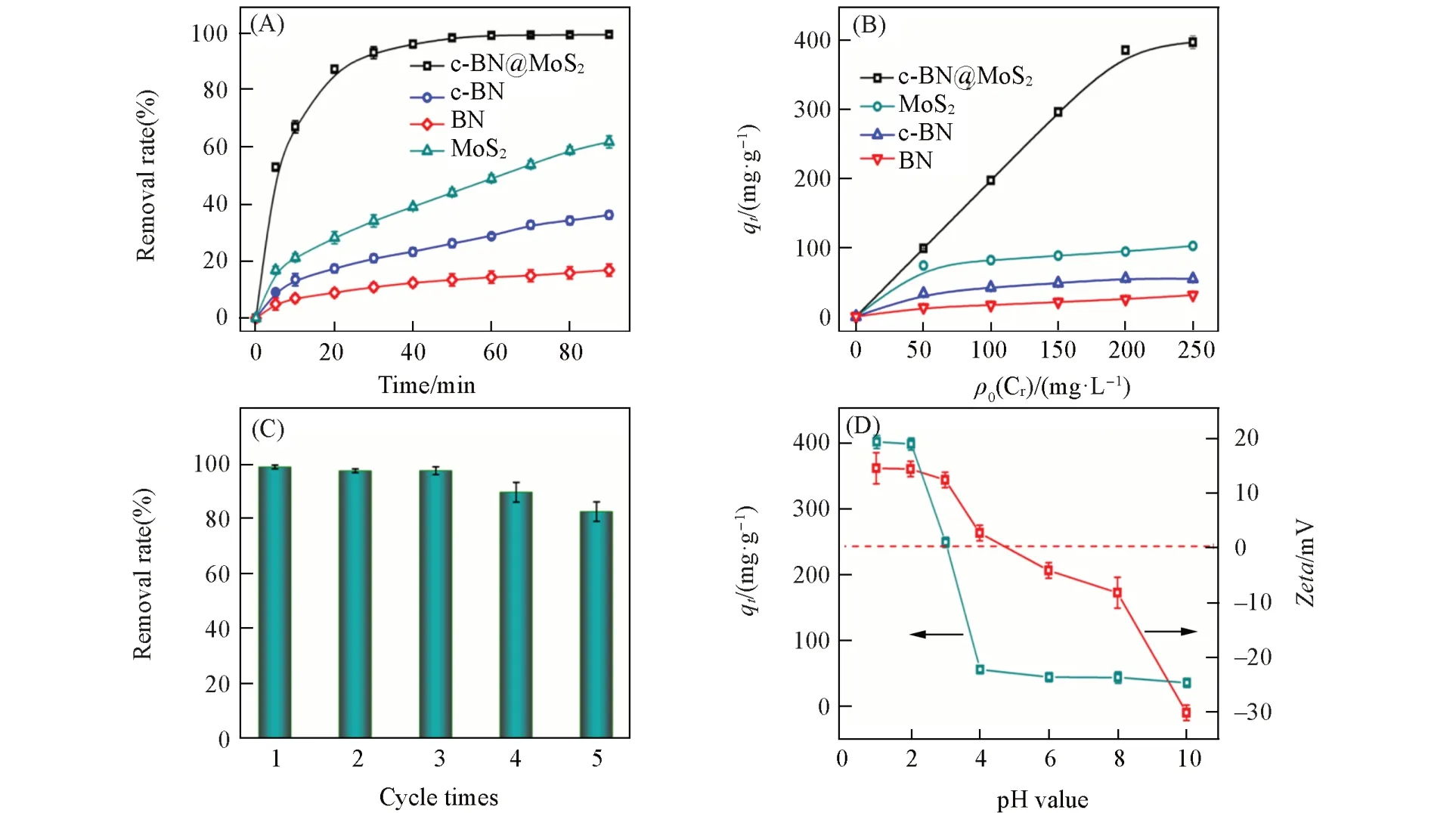
Fig.4 Removal rate(A)of Cr(Ⅵ)and removal capacity(B)in different systems,regeneration performance of c⁃BN@MoS2(C),removal capacity of Cr(Ⅵ)and Zeta potential at different pH values(D)
2.3 c⁃BN@MoS2还原去除Cr(Ⅵ)的机理
为了研究c-BN@MoS2去除Cr(Ⅵ)的过程,将c-BN@MoS2与不同浓度的Cr(Ⅵ)反应,反应后溶液中含有的Cr浓度如图5(A)所示.从图中可见,初始浓度低于150 mg/L,反应之后的Cr(Ⅵ)几乎被全部去除,然而溶液中还存在Cr(Ⅲ),其浓度分别为21.51,75.30和110.20 mg/L.当Cr(Ⅵ)的初始浓度为200,250 mg/L时,溶液中检测到总Cr浓度为159.90和187.01 mg/L,其中测试Cr(Ⅵ)的浓度分别为11.70,39.94 mg/L,由此计算含有Cr(Ⅲ)的浓度分别为148.20,147.69 mg/L.显然,反应总Cr含量低于反应前Cr(Ⅵ)的浓度,这说明c-BN@MoS2上还存在一部分Cr,为了进一步分析c-BN@MoS2上存在Cr的价态,将c-BN@MoS2与200 mg/L的Cr(Ⅵ)反应,对反应后的c-BN@MoS2进行XPS测试,Cr2p的窄谱图如图5(B)所示.可知,578.32和586.01 eV处出现的特征峰可属于Cr(Ⅵ)的Cr2p1/2和Cr2p3/2的结合能峰,577.01和587.50 eV处呈现的特征峰可归属于Cr(Ⅲ),其中Cr(Ⅵ)峰面积占72.53%,Cr(Ⅲ)峰面积占27.47%,这说明吸附到c-BN@MoS2上的Cr存在Cr(Ⅲ)和Cr(Ⅵ)两种价态,并以Cr(Ⅵ)为主.
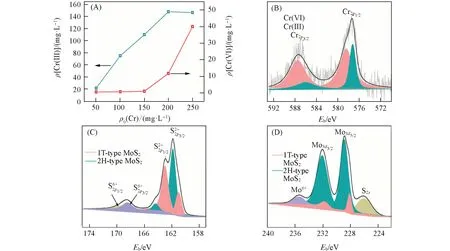
Fig.5 Cr(Ⅵ)and Cr(Ⅲ)content in solution after test(A),XPS spectra(B—D)after removal of chromium from c⁃BN@MoS2
为了进一步分析c-BN@MoS2吸附还原Cr(Ⅵ)的机理,首先对反应后的c-BN@MoS2进行了XRD测试,结果如图S6(见本文支持信息)所示.由图可知,去除Cr(Ⅵ)之后,c-BN@MoS2中MoS2的(002),(100)和(001)晶面对应的特征峰大幅下降,c-BN对应的特征峰强度也略有下降,这初步说明c-BN@MoS2中MoS2和c-BN均参与了反应.其次,采用XPS测试了c-BN@MoS2中S,Mo,N和B等元素结合能的变化.图5(C)是除铬后的S2p窄谱图,与除铬前的S2p窄谱图[图3(C)]相比,1T相MoS2中和分别从161.10和162.38 eV处偏移到160.98和162.98 eV处,且面积占比从60.88%降至10.82%;同时在168.45和169.27 eV处出现了新的特征峰,可分别属于,说明c-BN@MoS2中的S2−参与了还原Cr(Ⅵ)的反应[26],变成了S6+,产生了在反应后Mo3d的窄谱图[图5(D)]中,发现1T相MoS2的Mo3d5/2和Mo3d3/2的峰面积占比分别从23.91%,28.30%下降至5.16%和2.52%,而Mo6+的峰面积占比从5.30%增加至36.04%,这表明c-BN@MoS2中的Mo也参与了Cr(Ⅵ)的还原,Mo4+被氧化为Mo6+.将反应后的N1s窄谱图[见本文支持信息图S7(A)]与反应前[图3(A)]相比,N—B键、N—C键和吡啶N分别偏移到397.66,398.51和397.42 eV处,其中吡啶N峰面积占比从17.01%降低至10.52%,B1s的窄谱图[图S7(B)]中,B—N键和B—C键均发生偏移,B—C键面积占比从21.15%降低至8.92%,表明c-BN对Cr(Ⅵ)的去除起了重要作用.B—N键发生偏移,可能是由于BN中B空位和N空位对Cr(Ⅲ)和Cr(Ⅵ)进行了吸附[16].此外,c-BN表面上的碳可促进吡啶氮发生质子化产生正电荷,进一步通过静电吸引Cr(Ⅵ)阴离子[27].从上述结果可以推测c-BN@MoS2去除Cr(Ⅵ)的机理:c-BN@MoS2首先通过c-BN中B空位、N空位以及质子化的吡啶氮吸附溶液中Cr(Ⅵ),然后主要利用1T相MoS2还原约70%的Cr(Ⅵ)至Cr(Ⅲ),其中MoS2的S2-和Mo4+被氧化为S6+和Mo6+,而且被还原的Cr(Ⅲ)约88%进入反应溶液,仅有小部分Cr(Ⅲ)残留在c-BN@MoS2上.
2.4 c-BN@MoS2的催化性能及机理
SMX是一种治疗人和动物感染的代表性抗生素[28],会随着排泄物进入环境并对其造成危害.因此,将SMX作为催化对象来考察c-BN@MoS2作为助催化剂在Fe2+/PMS体系中的催化性能,结果如图6(A)所示.由图可知,反应10 min后,c-BN@MoS2/Fe2+/PMS体系对SMX的去除率可达99%,明显高于Fe2+/PMS体系(去除率38%)、MoS2/Fe2+/PMS体系(去除率67%)和c-BN/Fe2+/PMS体系(去除率77%),且反应4 min时的表观速率常数(Kobs)是Fe2+/PMS体系(表观速率常数0.15 min−1)的4.0倍、MoS2/Fe2+/PMS体系(表观速率常数0.22 min−1)的3.1倍及c-BN/Fe2+/PMS(表观速率常数0.3 min−1)的2.3倍.同时,为了考察c-BN@MoS2的重复使用性能,在降解完SMX后,对c-BN@MoS2离心,然后重新加入到SMX中,按照上述同样步骤重复6次,c-BN@MoS2对SMX的降解率仍能达到95%以上[图6(B)].SMX在c-BN/MoS2/Fe2+/PMS体系中反应10和20 min后,其TOC(总有机碳)去除率[图6(C)]分别达到43.11%和63.64%,说明SMX在c-BN@MoS2/Fe2+/PMS体系中具有较好的矿化效果,而且在反应30 min内,c-BN@MoS2/Fe2+/PMS体系也能有效降解磺胺嘧啶(SD)、双酚A(PBA)、苯酚(Phenol)、活性红195(3BF)和亚甲基蓝(MB)等污染物[图6(D)].
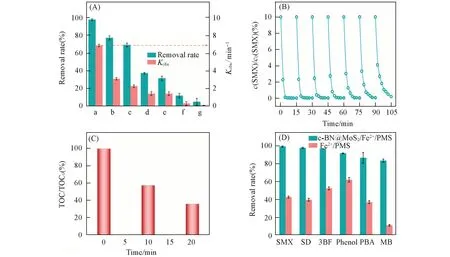
Fig.6 SMX removal under different systems and the corresponding Kobs values(A),reusability of c⁃BN@MoS2(B),TOC removal of SMX in the c⁃BN@MoS2/PMS/Fe2+(C)and removal of different pollutants in the c⁃BN@MoS2/Fe2+/PMS and Fe2+/PMS(D)
据文献[29,30]报道,羟基自由基(·OH)是Fe2+/PMS体系中生成的重要活性氧种,可以与甲醇(MA)和叔丁醇(TBA)发生反应,生成惰性中间产物.此外,硫酸根自由基(SO4·−)作为活性氧种也可与MA发生反应.因此用MA,TBA作为捕获剂研究c-BN@MoS2/Fe2+/PMS体系反应时存在的自由基,如图S8(见本文支持信息)所示.可见,随着MA,TBA的加入,明显抑制了反应进行,所以初步表明体系中产生了⋅OH和SO4·−.为了进一步考察c-BN@MoS2/Fe2+/PMS体系产生活性氧种的情况,通过EPR捕获技术来检测自由基[31],结果如图7(A)所示.可见,与Fe2+/PMS体系相比,c-BN@MoS2/Fe2+/PMS表现出了较高的⋅OH信号峰,信号峰几乎不变,上述分析表明c-BN@MoS2的加入主要促进了⋅OH的生成.
Fe3+到Fe2+的转化是快速活化PMS产生自由基的关键步骤[32].为了探究c-BN@MoS2促进Fe2+/PMS体系产生更多自由基的原因,测试了c-BN@MoS2/Fe2+/PMS体系和Fe2+/PMS体系中Fe3+和Fe2+浓度分别随反应时间的变化,结果分别如图7(B)和(C)所示.由图7(C)可知,在Fe2+/PMS中Fe2+的浓度连续下降,而在c-BN@MoS2/Fe2+/PMS中Fe2+的浓度[图7(B)]在5 min内下降到最低值,随后Fe2+的浓度逐步增大,15 min后趋于平衡,此时Fe2+含量为80%.与大多数助催化剂相比[图7(D)],Fe3+转变成Fe2+的反应速率和反应平衡时Fe2+含量占比均明显优于文献报道[33~36].显然,c-BN@MoS2的引入可以促使Fe3+向Fe2+转化.为了进一步分析c-BN@MoS2促进Fe3+向Fe2+转化的原因,采用XRD测试了c-BN@MoS2反应前后的变化,由图S6可知,c-BN@MoS2的结晶性减弱,MoS2和c-BN对应的特征峰均降低;采用XPS测试c-BN@MoS2中的Mo,N,B的结合能变化.图S9(A)(见本文支持信息)为Mo3d的窄谱图,由图可知,1T相MoS2的Mo3d3/2峰面积占比从28.30%降低到22.90%,属于2H相MoS2的Mo3d5/2峰面积占比从16.95%降低到3.95%,而在231.14 eV处拟合到新Mo6+的特征峰,可能是由于Mo4+与Fe3+发生了氧化还原反应.反应后N1s的窄谱图[图S9(B)]与反应前[图3(A)]相比,发现吡啶N面积占比从17.01%降低到7.96%,B—C键的峰面积比降低到5.19%,结合B1s的窄谱图[图S9(C)]中B—C键的峰面积比从21.15%降低到12.86%,说明c-BN对SMX的降解也做了一定贡献.基于上述分析,推测c-BN@MoS2增强Fe2+/PMS体系的机理如下:首先,Fe2+与PMS反应生成Fe3+⋅OH和SO4·−,然后Fe3+与c-BN@MoS2发生氧化还原反应,Fe3+从c-BN@MoS2得到电子转变成Fe2+,同时c-BN@MoS2中Mo4+被氧化为Mo6+,c-BN中石墨化碳和吡啶N可能提供电子使Fe3+和Mo6+分别还原为Fe2+,Mo4+,显然,c-BN@MoS2参与了Fe2+与PMS的反应,使整个反应得以循环进行.总之,c-BN@MoS2的存在大大加快了Fe2+/PMS体系中Fe3+向Fe2+的转变,有效解决了制约类芬顿技术催化效率的关键难点,导致更多·OH的产生,达到增强Fe2+/PMS体系去除抗生素污染物的目的.
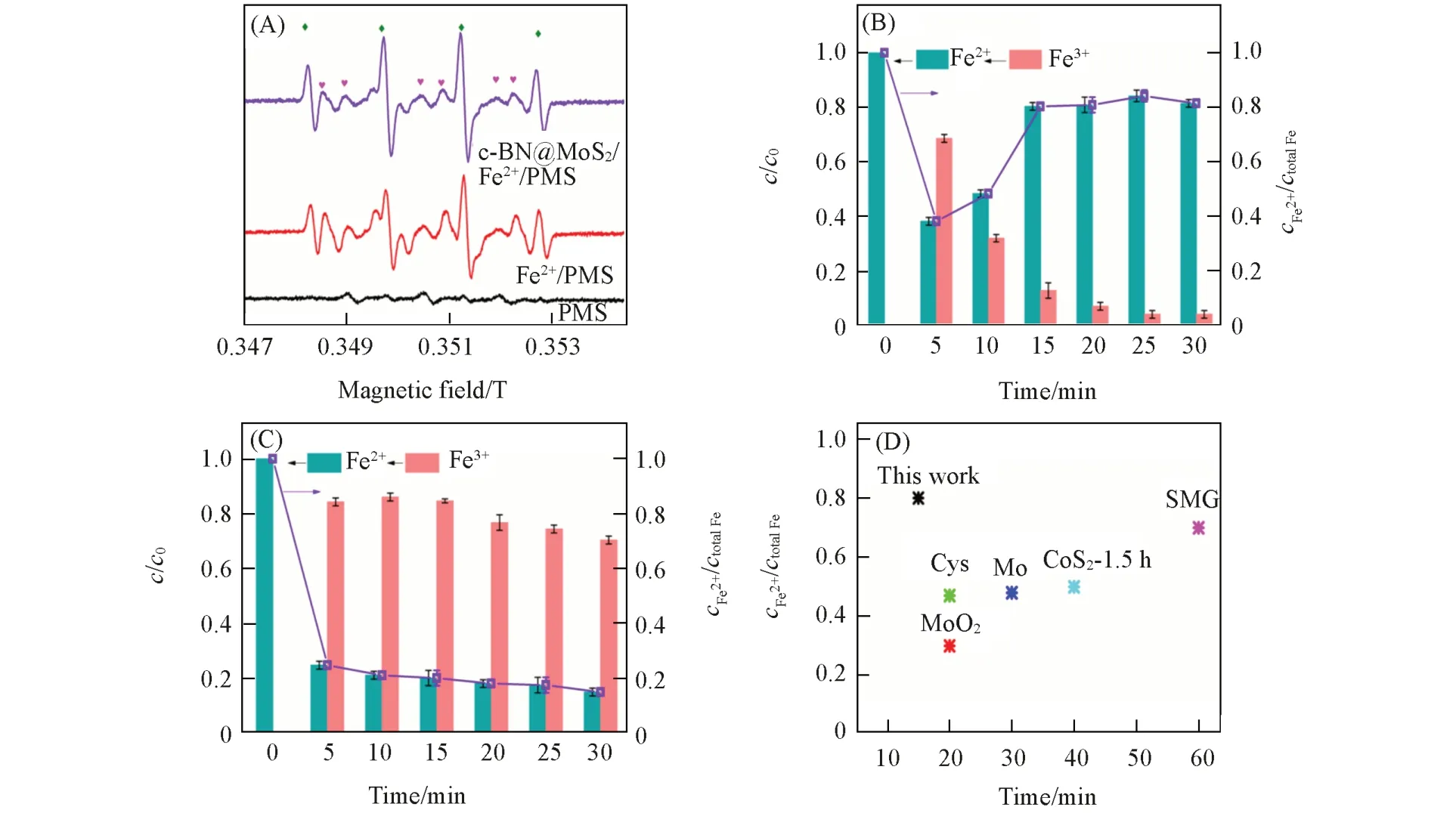
Fig.7 EPR spectra of DMPO⁃•OH and DMPO⁃SO4·-in different systems(A),the variation of Fe2+and Fe3+concentrations in the c⁃BN@MoS2/Fe2+/PMS(B)and PMS/Fe2+(C),concentrations of Fe2+in Fe2+/PMS system with different cocatalysts(D)
3 结 论
采用六方氮化硼作为MoS2的载体,结合多巴胺改性技术和水热法,制得一种碳掺杂六方氮化硼负载MoS2的纳米复合材料(c-BN@MoS2),研究了c-BN@MoS2对废水中Cr(Ⅵ)的去除能力和助催化降解有机污染物.研究结果表明,c-BN@MoS2对Cr(Ⅵ)具有优异的去除性能和再生性能,反应40 min时,对50 mg/L Cr(Ⅵ)的去除率达95%以上,且对Cr(Ⅵ)的最大去除容量可达401 mg/g,明显高于MoS2(98 mg/g),这主要归因于碳掺杂六方氮化硼的引入提高了MoS2的比表面积和平均孔径,这有利于吸附还原废水中的Cr(Ⅵ),更重要的是碳掺杂六方氮化硼促进了MoS2生成金属特性的1T相,促进了氧化还原过程中的电子转移.c-BN@MoS2作为Fe2+/PMS体系的助催化剂,反应10 min后SMX降解率高达99%,明显高于Fe2+/PMS体系(38%),而且反应速率常数为0.68 min−1,是单独Fe2+/PMS体系(0.15 min−1)的4倍,这主要是因为c-BN@MoS2的引入明显加快了Fe2+/PMS体系中Fe3+向Fe2+的转变,导致更多·OH的产生.研究结果为治理严重的重金属和抗生素污染废水提供了有益参考,而且也为制备高效去除重金属和有机污染物的双功能纳米复合材料提供了新思路.
支持信息见http://www.cjcu.edu.cn/CN/10.7503/cjcu20210429.
