甘松新酮/磷酸盐柱[6]芳烃主客体络合行为
2021-10-15苏丽娇李灿花鲁佳佳杨俊丽杨丽娟
杨 举,苏丽娇,李灿花,鲁佳佳,杨俊丽,古 捷,杨 丽,杨丽娟
(云南民族大学化学与环境学院,云南省智能超分子化学重点实验室,生物基材料绿色制备技术国家地方联合工程中心,昆明650500)
中药材甘松新酮(ND)性辛、甘、温,具有理气止痛、开郁解脾等功效.甘松新酮属于倍半萜类化合物,是干松属的活性成分[1].药理学研究结果表明,ND具有促进神经细胞生长、抗抑郁和镇静的生物学活性[2~7],其在临床上用于治疗心率失常,是抗心律失常常用药参松养心胶囊和稳心颗粒的主要成分[8,9].然而,甘松新酮因生物利用度低,水溶性较差,在一定程度上限制了其在药物制剂方面的应用[10].因此,寻找一种适宜的低毒药物载体显得尤为重要.
大环分子具有良好的主客体性能和生物兼容性[11,12],近年来被广泛应用于生物医用材料的构建[13].柱芳烃作为第五代经典大环分子,极大地丰富了大环化学的主客体系的研究.其中,水溶性柱芳烃作为一类重要的衍生化柱芳烃,具有亲水的边缘和疏水的刚性空腔,能容纳多种客体分子[14],近年来基于水溶性柱芳烃构筑的载药体系备受关注.2013年,Wang等[15]首次报道了水溶性柱[6]芳烃与米托蒽醌的载药体系.研究表明,在酸性条件下,该超分子体系可快速释放药物分子,并表现出较低的细胞毒性.该课题组[16]进一步选用羧基化柱[6]芳烃与偶氮苯化合物构筑了主客体络合体系,并用于药物米托蒽醌的包载与释放.Huang等[17]将水溶性柱芳烃引入智能超分子纳米转运体系,构筑了负载盐酸阿霉素的聚合物囊泡.研究表明,该囊泡不仅可以降低药物的毒性还能提高药效.以上研究为大环分子柱芳烃在药物包载、靶向递送和可控释放等方面的研究提供了巨大的应用前景,同时也表明水溶性柱芳烃作为天然药物的载体有一定的研究意义[18,19].
本课题组在前期工作中,以大环分子环糊精为药物载体,研究了环糊精与巴西木苏、鬼臼毒素、橙皮素和虫草素等天然药物分子之间的主客体络合行为[20~24].基于此,本文选用大环分子水溶性磷酸盐柱[6]芳烃(WP6P)作为主体(结构式如Scheme 1所示),ND作为客体,构筑了一种新型的ND/WP6P主客体包合物,并进一步研究了ND与WP6P的络合行为.研究结果表明,两者可形成稳定的1∶1络合物,其络合常数为5.160×104L/mol,且ND形成包合物后,其热稳定性也得到提高.这为ND在药物制剂方面的应用提供了理论依据,同时也为天然药物与大环分子的主客体行为研究提供了参考.

Scheme 1 Schematic diagram of inclusion of nardosinone(ND)and water-soluble phosphate salt pillar[6]aren(WP6P)
1 实验部分
1.1 仪器与试剂
三氟化硼乙醚、1,4-二(2-羟基乙氧基)苯、四溴化碳、三苯基膦、多聚甲醛、亚磷酸三乙酯、四甲基溴硅烷和氨水均为分析纯,上海泰坦科技有限公司;甘松新酮,纯度>98%(HPLC),陕西宝鸡晨光生物有限公司.
Aglient CARY ECLIPS型荧光分光光度计,安捷伦科技有限公司;Nicolet IS10型红外光谱仪(FTIR),美国Thermo科技有限公司;TTR 18 KW型转靶X射线衍射仪(XRD),日本理学公司,CuKα射线(λ=0.1546 nm),工作电流40 mA,工作电压40 kV,扫描速率5°/min,扫描区间3°~80°;Bruker Avance 400型核磁共振波谱仪(1H NMR和2D NMR,500 MHz),溶剂为D2O,瑞士布鲁克公司;NOVA NANOSEM-450型扫描电子显微镜(SEM),美国FEI公司;Agilent 8453型紫外-可见(UV-Vis)光谱仪,美国安捷伦公司;STA449F31型同步热分析仪(TG-DSC),N2气流速50 mL/min,升温速率10℃/min,升温范围25~500℃,德国耐驰公司.
1.2 实验过程
1.2.1 WP6P的合成参照文献[25]方法合成WP6P,合成路线如Scheme 2所示.对WP6P及中间体进行NMR表征,谱图数据与文献[25]报道一致.WP6P为黄色透明固体,产率98%.1H NMR(400 MHz,D2O),δ:6.88(s,12H),4.02(m,24H),3.46(s,12H),2.10(m,24H).
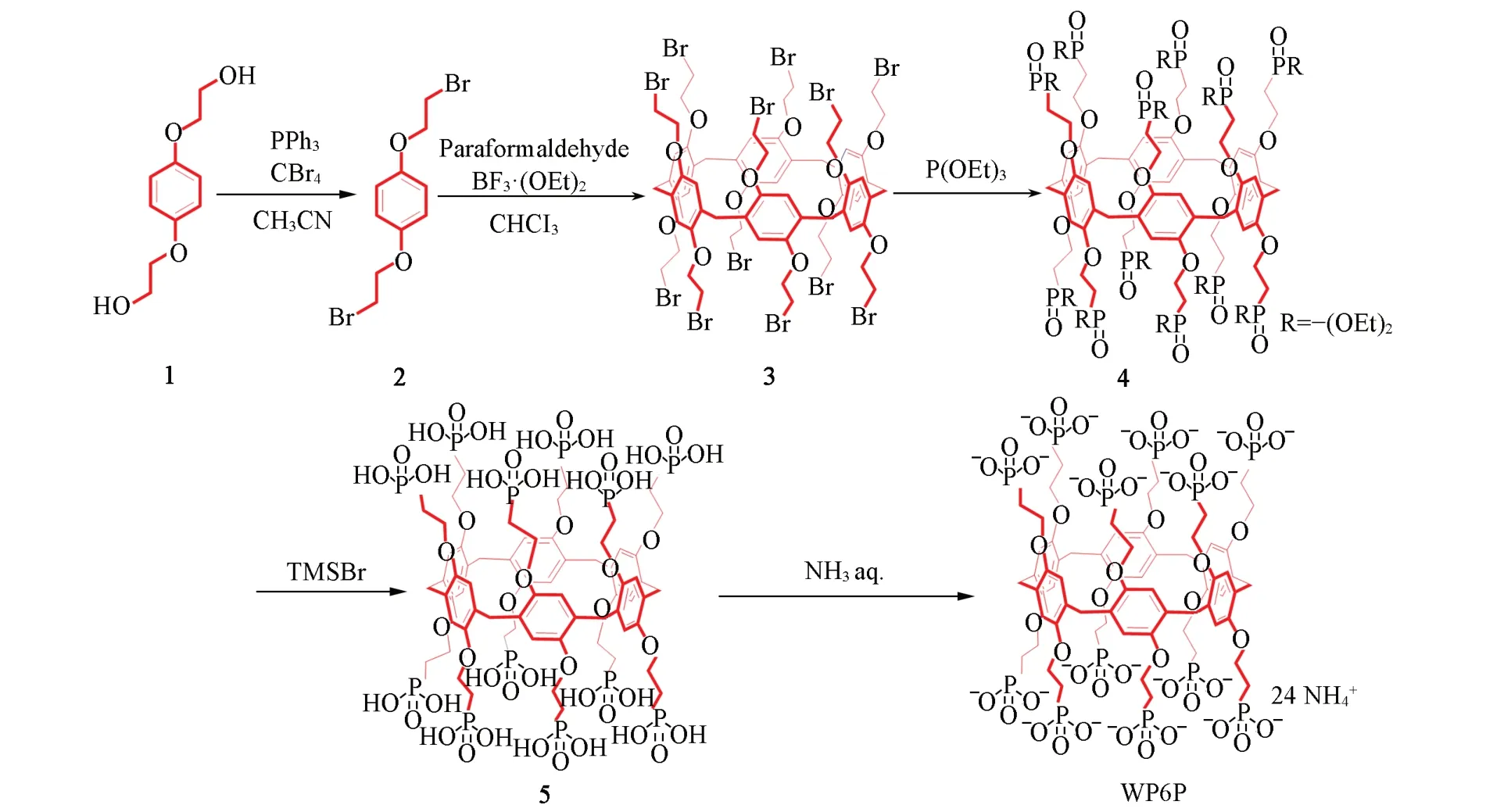
Scheme 2 Synthesis route of WP6P
1.2.2 ND/WP6P包合物及混合物的制备采用饱和溶液法制备ND/WP6P包合物.将0.0250 g(100 μmol)ND置于100 mL圆底烧瓶中,加入10 mL乙醇使其溶解;随后加入40 mL蒸馏水,充分混合均匀后,加入0.2298 g(100 μmol)主体WP6P,避光室温下搅拌24 h;将溶液用0.45 μm的微孔膜滤除不溶物,将滤液进行减压浓缩,于50℃下真空干燥,即得棕色固体包合物ND/WP6P.
准确称取0.0250 g(100 μmol)ND和0.2298 g(100 μmol)WP6P,搅拌使其充分混合均匀备用.
1.2.3 Job曲线的测定采用等摩尔连续变化法[26]测定ND与WP6P包合的化学计量比.保持ND与WP6P的总浓度为20 μmol/L,使ND与WP6P摩尔比在0~1内变化,分别配制一系列不同浓度的ND与WP6P混合溶液,摇匀后在最大吸收波长处测定各溶液的吸光度值.
1.2.4 紫外滴定实验通过紫外滴定实验研究了WP6P与ND的主客体络合行为及络合常数.保持客体ND的浓度不变(20 μmol/L),逐渐加入0,0.04,0.08,0.12,0.16,0.20,0.24,0.28和0.32 mL WP6P溶液(1×103μmol/L),用乙醇/水混合溶液(体积比1∶4)定容,摇匀后测定紫外光谱.
1.2.5 不同pH下ND与WP6P相互作用的紫外光谱测定为了进一步研究不同pH条件下ND与WP6的相互作用,测定了pH值分别为3.0和10.5条件下ND与WP6的紫外-可见吸收光谱.实验过程中维持ND的浓度不变(20 μmol/L),分别加入不同体积的WP6溶液(0,0.04,0.08,0.12,0.16,0.20,0.24和0.28 mL),用Na2HPO4-柠檬酸缓冲液和Na2CO3-NaHCO3缓冲液控制溶液的pH值为3.0和10.5,用乙醇/水混合溶液(体积比1∶4)和缓冲溶液混合定容,测定其紫外-可见吸收光谱.
1.2.6 分子对接为了进一步证实ND药物分子与WP6P发生了主客体作用,采用分子对接模拟了ND与WP6P的络合模式.由甲氧基柱芳烃经Gaussian View修饰得到WP6P的初始结构并进一步优化,甲氧基柱芳烃的结构取自剑桥晶体数据库,ND的结构通过Gaussian View构建.采用半经验PM3方法在无任何限制性因素的条件下优化二者结构至无虚频,将优化后的结构作为分子对接的输入构型[27].使用AutoDock 4.2程序,选用拉马克遗传算法(Lamarckian,GA 4.2)进行构象搜索500次,最大评估数为2.5×107,其它参数使用默认值,将ND对接到WP6P的空腔中.
2 结果与讨论
2.1 Job曲线的绘制
以ND的摩尔分数(xND,%)为横坐标,吸光度之差(ΔA)与xND之积为纵坐标,绘制Job曲线.图1给出ND与WP6P的Job曲线,可见ND与WP6P包合体系的吸光度之差在xND=0.5时达到最大值,说明ND与WP6P按照1∶1的化学计量比进行包合.
2.2 紫外滴定光谱分析
图2 (A)为ND与WP6P的紫外滴定光谱.根据浓度和吸光度的变化,作出ND和WP6P的浓度与吸光度的相关曲线图[图2(B)],采用Benesi-Hildebrand曲线法[28]求解得到ND与WP6P的络合常数为5.160×104L/mol,计算公式为1/(A−A0)=1/[(A∞−A0)K]×1/c+1/(A∞−A)[式中:A∞为主体浓度最大时的吸光度;A0为客体单独存在时的吸光度;c(μmol/L)为主体的浓度].络合常数K为5.160×104L/mol说明主客体之间存在较强的络合作用.
2.3 不同pH值下紫外滴定光谱分析
图3 为ND与WP6P在pH=3.0和10.5条件下的紫外滴定光谱.根据吸光度的变化,采用Benesi-Hildebrand曲线法[28]求解,得出ND与WP6P在pH=3.0和pH=10.5条件下的络合常数分别为7.196×104和5.747×104L/mol.可见,酸性条件下的络合常数较大,说明ND与WP6P在酸性条件下能形成较稳定的络合物.
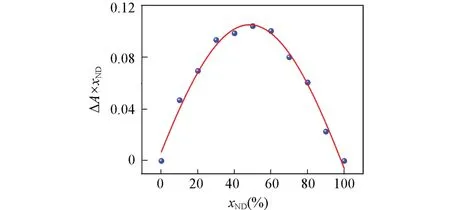
Fig.1 Job’s plot of ND/WP6P inclusion complex
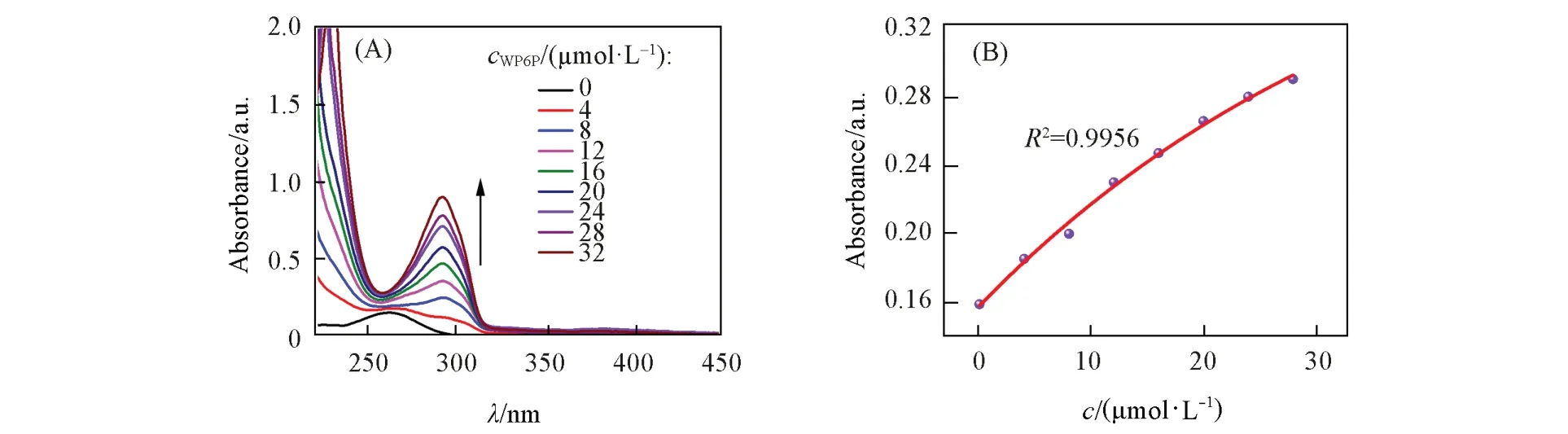
Fig.2 Ultraviolet titration curves of WP6P solutions of 0—32 μmol/Ladded in turn to the ND solution of 20 μmol/L(A)and the relationship between the absorbance of ND and WP6P concentration(B)
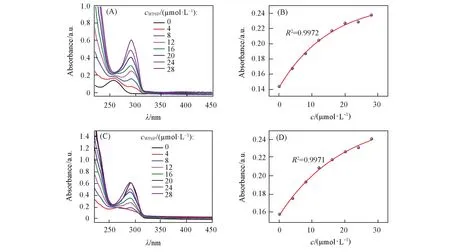
Fig.3 Ultraviolet titration curves of WP6P solutions of 0—28 μmol/L added in turn to the ND solution of 20 μmol/L at pH=3.0(A),pH=10.5(C)and the relationship between the absorbance of ND and WP6P concentration at pH=3.0(B),pH=10.5(D)
2.4 SEM分析
图4 示出了ND,WP6P,ND/WP6P物理混合物及ND/WP6P络合物的SEM照片.可见,ND为表面光滑的块状结构,WP6P为细碎的片层结构;二者的物理混合物是WP6P的碎片均匀地堆积在ND的表面;而络合物则呈现出全新的微观形貌,表明主客体之间形成了络合物.

Fig.4 SEM micrographs of ND(A),WP6P(B),ND/WP6P physical mixture(C)and ND/WP6P inclusion complex(D)
2.5 红外光谱分析
图5 示出了主客体络合前后红外光谱吸收峰位置和强度的变化情况.由图5谱线a可见,2983.46和2939.11 cm-1处为ND结构中—CH2的伸缩振动吸收峰,1686.53 cm-1处为C=O的伸缩振动吸收峰,1615.16和1311.41 cm−1处分别为C=C和C—O的吸收峰.由图5谱线b可见,1644.09和1499.45 cm−1处为苯环上的C=C骨架伸缩振动峰,1211.13和1125.31 cm−1处为P=O的伸缩振动峰,897.74,798.42和537.10 cm−1处为苯环上C—H的面外弯曲振动峰.由图5谱线c和d可见,物理混合物(ND/WP6P)在1311.41和1272.84 cm−1处明显存在ND上碳氧单键的特征峰,而络合物中并不明显,且在络合物中2983.46和2939.11 cm−1处—CH2的伸缩振动吸收峰消失.ND与WP6P络合前后红外光谱的变化说明客体药物分子ND与WP6P大环主体形成了主客体络合物.
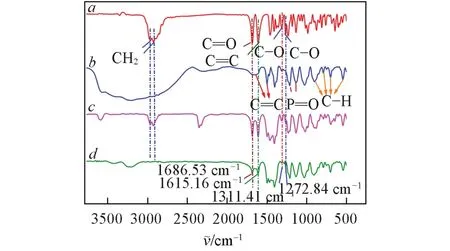
Fig.5 FTIR spectra of ND(a)WP6P(b),ND/WP6P physical mixture(c)and D/WP6P inclusion complex(d)
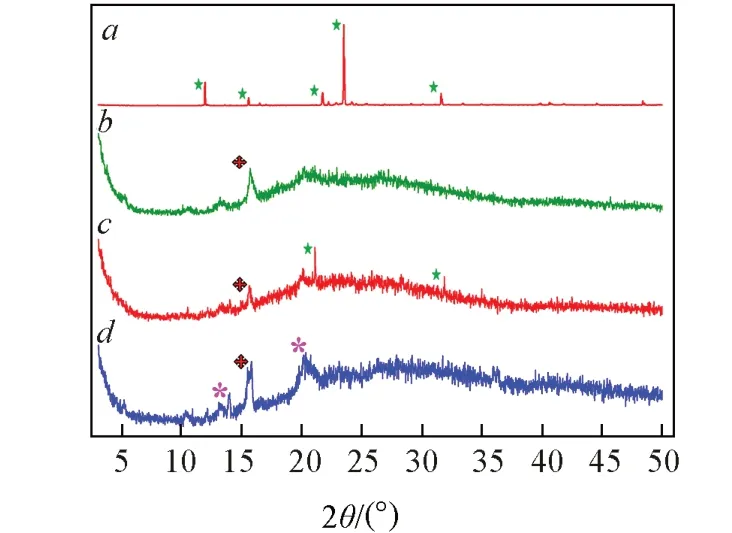
Fig.6 XRD patterns of ND(a),WP6P(b),ND/WP6P physical mixture(c)and ND/WP6P inclusion complex(d)
2.6 X射线粉末衍射分析
图6 为ND,WP6P,ND/WP6P物理混合物及ND/WP6P络合物的XRD谱图,WP6P呈现出明显的无定形结构,在2θ=15.09°~16.95°范围内有一个较宽的衍射峰.ND呈现出晶型结构,在2θ=11.89°,15.54°,21.71°,23.45°,31.56°处均有较强且尖锐的衍射峰.物理混合物的XRD衍射谱线上仍然有ND的衍射峰,表明ND与WP6P是简单的叠加.当ND与WP6P形成络合物后,其衍射模式与WP6P的衍射模式类似,呈现出一个较宽衍射峰的无定形结构,在2θ=13.86°处出现了新的衍射峰,在2θ=15.72°处的衍射峰强度明显增强,在2θ=19.17°~21.83°处出现了较宽的衍射峰.ND与WP6P形成络合物后,某种程度上二者结构重新排列,比单一的ND具有更显著的不定形结构,说明ND与WP6P形成了主客体络合物.
2.7 热重分析
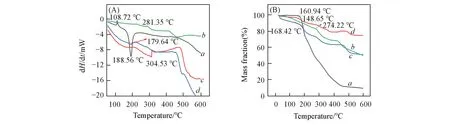
Fig.7 DSC(A)and TG(B)curves of ND(a),WP6P(b),ND/WP6P physical mixture(c),and ND/WP6P inclusion complex(d)
由图7(A)所示DSC曲线可见,ND在108.72℃有一个向上的融化时的吸热峰,在188.56℃有一尖锐的向下的放热峰;WP6P的吸热峰则出现在281.35℃处.在二者的物理混合物中可观察到ND在188.56℃处的放热峰和WP6P的放热峰,说明简单的物理混合并未使二者的热力学性质发生明显的变化.在形成包合物后,无论是WP6P还是ND,自身的吸热峰均消失,说明包合物中已没有自由态的ND分子,且在304.53℃处出现了新的放热峰,进一步验证了已成功制备该包合物.由图7(B)所示TG曲线可见,ND,WP6P,物理混合物和包合物在160.94,274.22,148.65和168.42℃出现了拐点,说明此时物质开始分解.具体表现为ND在开始时分解缓慢,出现拐点时仅失重2%,WP6P失重18%,物理混合物失重13%,包合物则失重4%,但在200℃后包合物的失重较为缓慢,说明该包合物热力学性质发生了改变,热稳定性得到了提高.
2.8 一维核磁共振氢谱分析
通过1H NMR研究了主客体络合前后化学位移的变化,进一步分析二者的络合情况[29].图8为ND/WP6P包合物的1H NMR谱图.可见,在δ0.88~1.38之间和δ3.44处出现了ND的特征质子峰,且包合前后WP6P特征质子峰的化学位移发生了变化,其变化值(ΔδH)见表1.WP6P的H1,H2和H3质子信号的化学位移均向高场发生了移动,ΔδH分别为0.04,0.01和0.32;而H4的化学位移则向低场发生明显移动,△δH为0.02.进一步证明形成了ND/WP6P包合物.
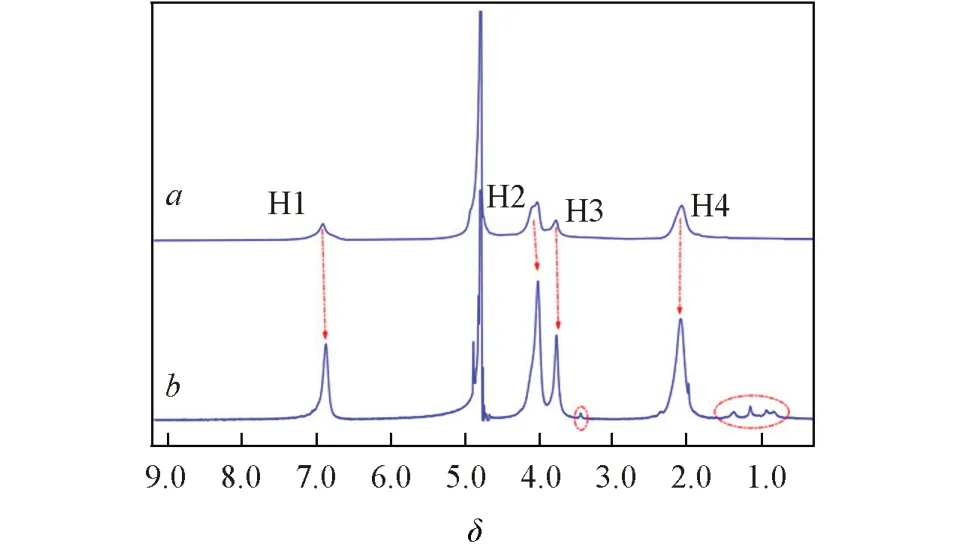
Fig.8 1H NMR spectra of WP6P(a)and ND/WP6P inclusion complex(b)(D2O,25℃)
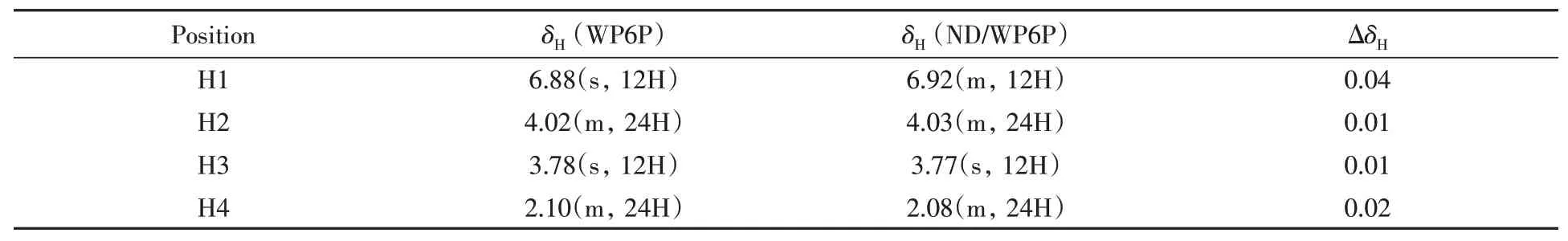
Table 1 Chemical shift values(δH)of WP6P and ND/WP6P inclusion complex andΔδH of WP6A before and after inclusion
2.9 二维核磁共振氢谱分析
主客体之间的包合模式可以通过二维旋转坐标系豪泽效应谱(2D ROESY)来推测[30].由图9(A)可见,ND的H4,H7,H14,H15与WP6P的H4(苯环对位O—C键碳上的质子)存在相关.由于WP6P的H4位于柱芳烃两端的支链上,而ND与两端的H4都存在相关,结合络合物的一维核磁共振氢谱数据的分析结果,可推断出ND与WP6P的主客体包合模式如图9(B)所示.
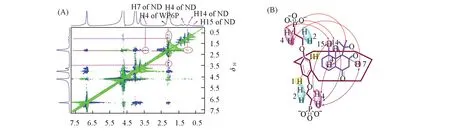
Fig.9 2D ROESY spectra of ND/WP6P inclusion compound(D2O,25℃)(A)and possible inclusion mode and key NOEY correlations(B)
2.10 分子对接模拟
通过500次的构象搜索,获得了不同的能量构象.由于结合能越低,亲和力越大,则构象也越稳定,经过对构象搜索结果分析,得到其最低结合能的包合模式(图10),即客体ND处于WP6P的空腔中,并与其形成稳定的包合物.结果表明,主客体间未形成氢键,包合物形成的驱动力主要为疏水作用.
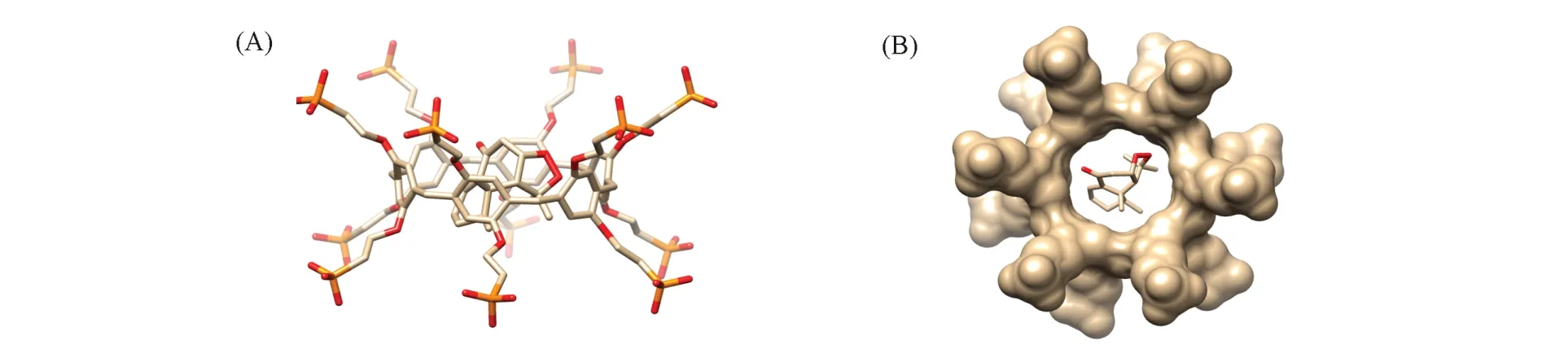
Fig.10 Front view(A)and top view(B)of optimal inclusion mode of ND/WP6P
3 结 论
采用简便、绿色、无污染的方法制备了甘松新酮/磷酸盐柱[6]芳烃(ND/WP6P)主客体包合物.利用紫外光谱滴定实验研究了二者的主客体络合行为,结果表明,ND与WP6P能形成摩尔比1∶1的包合物,其结合常数为5.160×104L/mol.此外,通过红外光谱和X射线粉末衍射、扫描电子显微镜以及热重分析对包合物进行了表征,结果表明,ND/WP6P包合物制备成功,且热稳定性得到明显提高.采用核磁共振波谱、分子对接模拟等手段分析了ND与WP6P之间的包合模式,结果表明,主客体间没有氢键形成,包合物形成的驱动力主要为疏水作用.本文研究结果可为柱芳烃在药剂学方面的应用提供一定的理论依据和参考.
