2,6,9-三取代嘌呤衍生物三维定量构效关系和靶点相互作用
2021-10-13朱财盛麦曦冯丽华杜巧丽何玲何卫保张其民
贾 磊,朱财盛,2,麦曦*,冯丽华,杜巧丽,何玲,何卫保,张其民
(1.南昌大学药学院,江西 南昌 330006;2.中国科学院上海药物研究所,上海 201203)
嘌呤是一类重要的生命活性物质,参与生物体内的新陈代谢过程、生命过程中的能量转移、核酸合成以及多种生化反应,具有广泛的生物活性,对嘌呤进行结构修饰改造后能获得抗肿瘤、抗病毒及抗炎等多种活性的药物[1],如抗代谢物巯嘌呤和巯鸟嘌呤是传统的抗肿瘤药物[2-3],嘌呤类核苷药物奈拉滨、氟达拉滨、克拉利滨和克罗拉滨等均为广泛用于临床的癌症治疗药物[4-5]。
对嘌呤母核的2,6,8,9位进行结构修饰发现了多种作用机制的嘌呤类衍生物,主要有细胞周期蛋白依赖性激酶抑制剂(cyclin dependent kinase,CDKs)[6-7]、热休克蛋白90(Hsp90)抑制剂及DNA拓扑异构酶抑制剂等;其中的N,N-二甲基腺嘌呤(6-DMAP)是最早应用于临床的嘌呤类CDKs抑制剂,而后进一步筛选得到Olomoucine,其对CDK1(IC50=7.0 μmol·L-1)、CDK2(IC50=7.0 μmol·L-1)和CDK5(IC50=3.0 μmol·L-1)都有抑制作用,对Olomoucine进行结构改造得到Olomoucine II,发现其对CDK1抑制活性显著提高(IC50=0.1 μmol·L-1)[8]。Bukanov等[9]通过对Olomoucine II进行结构优化,设计合成出化合物R-CR8(IC50约为19.0 nmol·L-1)。Wu等[10]发现嘌呤类CDKs抑制剂Seliciclib对CDK1、CDK2、CDK5、CDK7都有很强的抑制作用,现已进入多项II期临床研究。MPC-3100是Myriad Pharmaceutiicals Inc公司研发的嘌呤类Hsp90抑制剂,并于2011年完成了I期临床试验[11];另一个嘌呤类Hsp90抑制剂Debio 0932已进入临床II期试验,其主要针对非小细胞肺癌的治疗[12]。Barbara等[13]采用两阶段虚拟筛选的方法,分别从已建立的9-H嘌呤类和1H-吡唑并[3,4]嘧啶类拓扑异构酶IIa抑制剂中筛选出具有微摩尔级别抑制活性的新化合物,发现化合物13和22对人肝癌细胞(HepG2)和人乳腺癌细胞(MCF-7)显示出良好的抗增殖活性。
定量构效关系(Quantitative Structure-Activity Relationship,QSAR)是一种借助化合物的理化参数或结构参数,以数学和统计学手段定量研究化合物结构与生物活性、化合物在生物体内吸收、分布、代谢、排泄等相关性质的方法。常用的方法有二维QSAR(2D-QSAR)和三维QSAR(3D-QSAR)方法,2D-QSAR通过计算分子的物理化学参数,建立理化参数与活性之间的线性模型或非线性模型后,用于新化合物的活性预测、筛选及设计[14]。3D-QSAR是建立在分子的三维结构与各类活性之间的关系,通过力场计算,建立分子体积、分子形状、电荷分布与活性之间的关系,代表性的3D-QSAR方法有比较分子场分析法(Comparative Molecular Field Analysis,CoMFA)[15]与比较相似性指数分析法(Comparative Molecular Similarity Indices Analysis,CoMSIA)[16]等。易位体比较分子场法(Topomer CoMFA)是由Cramer等[17]基于二维片段分析开发的新型3D-QSAR工具,Topomer CoMFA模型是将化合物分子结构切割成若干个碎片,运用靶点筛选技术,用其他基团或者分子结构替换原分子结构中的某些碎片,利用搜集的活性数据进行分析,建立的模型可以基于原碎片的虚拟筛选以及对分子的官能团机构进行替换优化。与CoMFA和CoMSIA不同的是,Topomer CoMFA是通过叠合替换规则完成的3D-QSAR准备工作,具有重复性高的优势,利用Topomer CoMFA可快速建立模型并进行分析与评价,为构建构效关系提供有利的理论依据。本文拟采用Topomer CoMFA研究2,6,9-三取代嘌呤类衍生物的3D-QSAR,并采用分子对接Surflex-dock法研究嘌呤类衍生物与CDK2的作用模式和作用机制,分析影响嘌呤类衍生物抗肿瘤活性的分子结构特征,建立具有显著预测能力的嘌呤类化合物活性预测模型,为创新药物的研究提供科学依据。
1 实验部分
1.1 数据集来源和结构构建
本论文合成了45个2,6,9-三取代嘌呤类衍生物,,结构通式如图1所示用MTT法测定所有化合物对非小细胞肺癌细胞A549的生长抑制作用,计算各化合物对肿瘤细胞生长抑制率,采用改良寇式法计算各化合物的半数抑制浓度IC50值(μmol·L-1),所有IC50值均将单位转化为mol·L-1,然后均转化为负对数pIC50(=-lgIC50)。用SYBYL进行3D-QSAR建模和分子对接,使用Sketch Molecule模块构建了所有化合物图像的3D结构(表1和图1),利用Tripos力场和Gasteiger-Huckel电荷优化每个分子(创建训练集,测试集和分子对接),使用Powell梯度算法进行结构能量最小化,其收敛准则为0.005 kcal·mol-1,最大值为10 000次迭代。
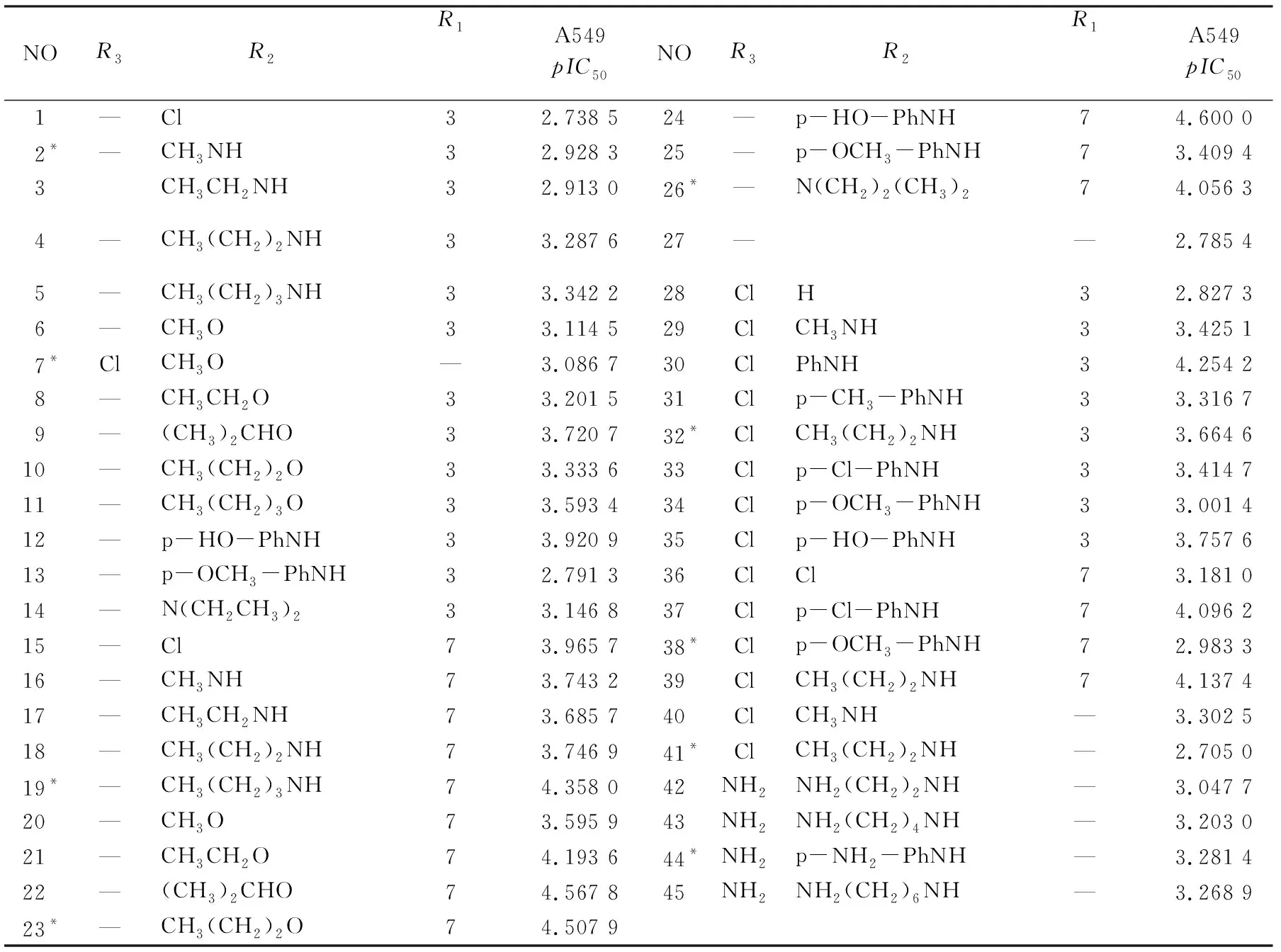
表1 嘌呤衍生物分子结构和活性值a)
1.2 Topomer CoMFA模型的构建
将45个化合物随机分组,36个化合物组成训练集(80%),9个化合物组成测试集(20%)。采用Topomer技术将化合物分子切割成几个片段,并生成碎片三维结构,碎片根据一定的经验规则进行调整后,进行Topomer CoMFA分析,计算分子的电性参数和立体参数,以电性和立体参数为自变量,pIC50为因变量,采用偏最小二乘法(Partial least squares,PLS)进行模型拟合,抽一法(Leave-One-Out)交互验证检验模型的内部预测能力,测试集验证模型的外部预测能力。
1.3 分子对接
采用Surflex-dock研究化合物与CDK2的作用模式和作用机制,CDK2三维晶体结构来源于PDB数据库(http://www.rcsb.org/,登录号1HCK),为三磷酸腺苷(ATP)活性位点共晶结构,经提取ATP配体、去水、加氢等修饰后,以配体方式产生活性位点,设置Surflex-dock配体输出构象个数为20个,其余参数默认,以原配体结构作参照对比,进行化合物与CDK2活性位点的对接及打分,以Total score为输出构象的总打分函数。
2 结果与讨论
2.1 Topomer CoMFA建模结果和评价
Topomer CoMFA建模过程中,断裂方式的选择对模型的质量影响较大,本研究中的嘌呤衍生物主要以6,9位取代为主,因此采用图1所示对A键和B键进行3种方式的Topomer切割断裂后,将化合物分为2个或3个片段,计算各片段的电性参数和立体参数,采用PLS通过交叉验证得到最佳主成分数n后进行非交叉验证回归,建立了3个3D-QSAR模型(表2);同时,得到交叉验证系数Q2和非交叉验证相关系数r2,预测值的标准误差SD以及显著性检验F。交叉验证系数Q2越大,相关系数r2越大,SD值越小,表示相关性越好,模型的预测能力越强。而F主要用于判断样本间是否有显著差别,其值越大,说明差别越显著。对于TopomerCoMFA模型,一般Q2>0.5,r2>0.6,F>100,即表明所建模型具有较好的统计学意义和预测能力。

表2 Topomer CoMFA模型验证结果a)
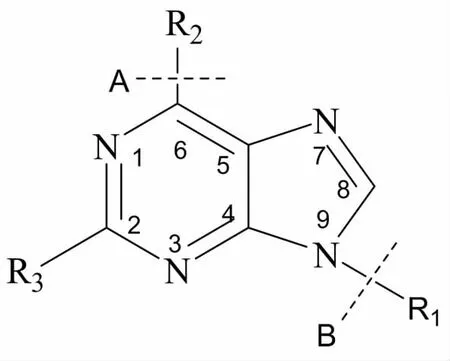
图1 嘌呤衍生物结构及断裂方式

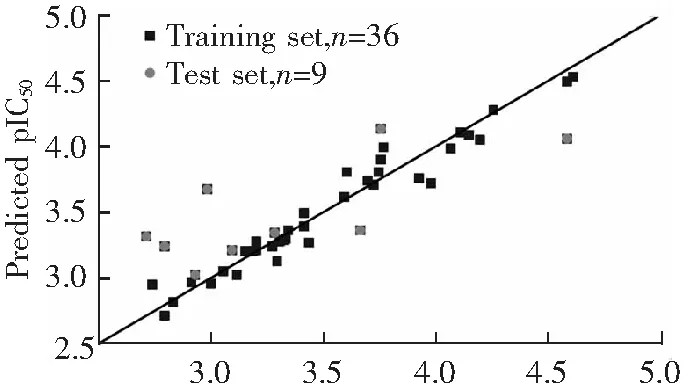
Experimental pIC50

2.2 构效关系分析
化合物3结构较简单,以该化合物为样本分子做3D-QSAR分析。如图3(A)所示,化合物3按表2模型1的的断裂方式切割为Ra及Rb二个分子片段,图3(B)和图3(D)分别为Ra及Rb的立体场三维等势图,图中绿色区域表示此处引入体积较大的取代基有利于活性的提高,而黄色区域表示此区域不宜有大体积的取代基,在图3(B)中,嘌呤8位附近聚集了较大体积黄色等势域,表明8位不宜引入取代基;9位取代基的中段和远端聚集了大片绿色等势域,表明引入较大体积取代基有利于化合物活性提高,如9位取代基为n=7碳链取代的化合物15,16,17,18,19,20,21,22,23,24,25的活性均高于相应的n=3碳链取代的相应化合物1,2,3,4,5,6,8,10,11,12,13,14,尤其是化合物24为活性最高的化合物;图3(D)中,嘌呤6位取代基甲氨基处有体积较大的绿色区域,表明嘌呤6位可以引入体积较大取代基,因此用体积更大的丙胺和丁胺取代后生成的化合物4和5的活性逐渐提高。
图3(C)和图3(E)分别为Ra及Rb的静电场三维等势图,图中红色等势域表示引入负电性基团有利于化合物活性的增加,而蓝色等势域表示此区域可引入正电性基团;由图3(C)可知,嘌呤2位取代基周围被红色等势域所包围,表明嘌呤2位宜引入负电性取代基,在本类化合物中,化合物28~44均为2位具有负电性氯取代基的衍生物,它们的活性与相应没有氯取代的化合物相比有部分化合物活性有增加,如28,29,34分子与相对应在2位无氯取代基的1,2,13相比均增加了活性;但有的化合物引入氯取代基后活性降低了,如36,38,39分子与无氯取代基的15,25,18相比,活性均有所下降;这种现象与嘌呤环中杂原子氮对氯取代基影响有关,在鲍林电负性中氯的电负性大于氮的电负性,而在阿莱-罗周的电负性中却是氮的电负性大于氯的电负性,即氮和氯的电负性是近似的,因此嘌呤2位的取代氯虽然为负电性,但对活性的影响并无规律性。由图3(E)可知,Rb的近端和中端为红色和蓝色等势域交错,表明此区域可引入一些极性取代基,远端为蓝色等势域,表明远端宜引入带正电取代基,此结果表明在嘌呤6位宜引入一些极性基团。
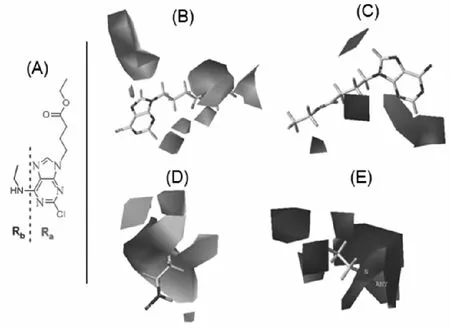
(A)化合物3的断裂方式;(B)Ra的立体场等势图;(C)Ra的静电场等势图;(D)Rb的立体场等势图;(E)Rb的静电场等势图。
2.3 与靶点作用模式
肿瘤细胞中的细胞周期调控缺陷由CDKs/Cyclin失调直接或间接导致,即与Cyclin和CDKs过量表达或活性异常增强有关[19]。由于CDKs在调控肿瘤细胞增殖与凋亡中起关键作用,抑制CDKs可有效阻止癌细胞周期进程,从而抑制肿瘤细胞增殖[20],研究嘌呤类衍生物对CDKs的作用模式可以探究其抗肿瘤作用机制,为其结构改造提供依据,本文采用Surflex-dock分子对接法研究了本类嘌呤衍生物与ATP的活性位点CDK2的作用模式与机制,ATP的活性位点共晶结构的PDB号为1HCK,是研究得最为深入的一个CDK亚型结构[21-22],其活性结构主要分为绞链区、核糖/磷酸盐结合区和Phe80口袋区,绞链区(残基81-84)存在着一些氢键供体和受体的结合位点,其中最关键的为Leu83和Glu81,ATP的腺嘌呤环能够与Leu83和Glu81同时形成氢键,一些强效的抑制剂CDKs抑制剂,如R-roscovitine、Purvalanol B和Olmoucine等都能与Leu83的羰基之间形成氢键[23];CDKs结构中有一个由Phe80形成的浅腔,虽然没有被ATP占据,但是在一些抑制剂中都将疏水环伸向了该位点[22];ATP中的磷酸基团能与CDKs晶体结构中的Lys33、Asp145作用,这一区域具有高度的亲水性和柔性。对化合物进行设计时将该亲水性区域加以考虑可能对提高化合物的活性和选择性有帮助。图4(A)和4(B)分别为高活性化合物24和低活性化合物1的氢键作用图,化合物24嘌呤环的6位芳环取代基的羟基能够与绞链区的Leu83和Glu81形成2个氢键,嘌呤环的5位N与核糖/磷酸盐结合区的Lys33形成1个氢键,9-位长侧链的酯基氧与Gln131形成第1个氢键;而化合物1仅形成2个氢键,分别为5位N与Asn132及9位侧链酰基氧与Lys89形成,化合物1没有与关键的氨基酸残基Leu83和Glu81形成氢键,或许是其活性低于化合物24的原因之一,同时也与其6位仅为氯取代基有关,该结果表明在嘌呤6位宜引入能形成氢键的基团。图4(C)和4(D)是为化合物24和化合物1在CDK2活性口袋中的键合模式图,化合物24将嘌呤6-位取代的芳环伸向Phe80和Phe82形成的疏水腔,9位取代侧链伸向Asp145处的亲水腔;而化合物1则是将嘌呤9-位取代侧链伸向疏水腔,6-位取代氯伸向亲水腔。
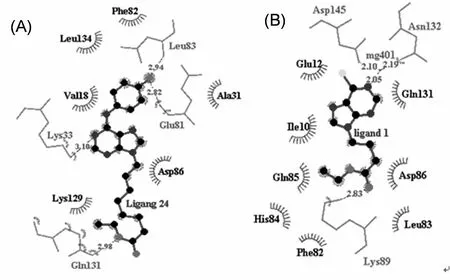

图4 化合物24(A)(C)和化合物1(B)(D)与CDK2键合模式图(绿色虚线代表氢键)
3 结论
本文通过Topomer CoMFA方法成功建立了2,6,9-三取代嘌呤类衍生物的3D-QSAR模型,其交叉验证系数为0.929,F检验值为100.35,外部交叉验证系数为0.566,模型具有优良的预测能力;3D-QSAR模型表明,嘌呤9-位引入较大体积取代基有利于化合物活性提高;嘌呤2-位宜引入一些负电性取代基;2,6,9-三取代嘌呤类衍生物与CDK2靶点的作用模式进一步表明,嘌呤6-位如引入能形成氢键的取代基将有利于与CDK2绞链区的关键氨基酸残基Leu83和Glu81形成氢键,因此综合3D-QSAR模型和分子对接研究结果,嘌呤6-位引入较大体积并能形成氢键的取代基将有利于化合物活性的提高。
