中国汉族心肌病患者FLNC 基因变异的家系研究
2021-10-13李雪洁周年伟孙敏敏汪咏莳谢惠琳舒先红
李雪洁 周年伟 孙敏敏 李 伟 汪咏莳 谢惠琳 舒先红
(1复旦大学附属中山医院心超室,2心内科 上海 200032;3上海市影像医学研究所 上海 200032;4上海市心血管病研究所 上海 200032)
FLNC编码的细丝蛋白C(filamin C,FLNC)是一种广泛存在于心肌及骨骼肌中的蛋白质,以同源二聚体的形式表达,主要通过与肌动蛋白交联维持肌节的机械稳定性。最初,FLNC基因突变在肌原纤维肌病中被发现,近年来在肥厚型心肌病(hypertrophic cardiomyopathy,HCM)、扩张型心肌病(dilated cardiomyopathy,DCM)、限制型心肌病(restrictive cardiomyopathy,RCM)和致心律失常性心肌病(arrhythmogenic ventricular cardiomyopathy,ACM)中也被陆续报道。随着基因筛查在临床中的应用,FLNC已经成为遗传性心脏病的常见致病基因之一,在HCM 和RCM 患者中发现了许多FLNC的错义突变,DCM 及ACM 患者中发现的则多为截断突变,这表明不同类型的FLNC突变可能存在不同的致病机制。然而中国汉族人群心肌病患者携带FLNC突变的变异谱仍不明确,其基因型-表型的关联性也值得进一步探究和讨论。
资料和方法
入组人群2018 年至2020 年就诊于复旦大学附属中山医院的150 名无血缘关系的汉族HCM 患者和50 名DCM 患者被纳入研究。成年人HCM 的临床诊断标准与以往文献一致:一个或多个左室心肌节段的最大室壁厚度≥15 mm,或有明确家族史者最大室壁厚度≥13 mm(排除后负荷增加),通常不伴有左室扩大;家族性HCM 定义为在同一家系中有两名以上的HCM 患者。DCM 的临床诊断标准为具有心室扩大和心肌收缩功能降低的客观证据:左室舒张末内径(left ventricular end-diastolic dimension,LVEDd)>5.0 cm(女性)和 LVEDd>5.5 cm(男性)(或>年龄和体表面积预测值的117%,即预测值的2 倍SD+5%);左室射血分数(left ventricular ejection fractions,LVEF)<45% 和左室短轴缩短率(left ventricular fraction shortening,LVFS)<25%,除外冠心病、高血压病、心脏瓣膜病、先天性心脏病、系统性疾病等;家族性DCM 定义为同一家系中有2 名或2 名以上的DCM 患者。收集患者的临床信息,包括发病年龄、确诊年龄、性别、症状、是否植入起搏器,并常规进行心电图(electrocardiogram,ECG)和 经 胸 超 声 心 动 图(transthoracic echocardiography,TTE)检查。该实验方案经过复旦大学附属中山医院人类伦理委员会批准[B2016-016(2)R],所有纳入本研究的患者均已签署知情同意书。
全外显子组测序及变异解释提取入组患者的外周血基因组DNA,在HiSeq XTEN 测序平台(美国Illumina 公司)上进行全外显子组测序。具体测序方法详见本课题之前的研究[1-2]。测序后使用GATK(Genome Analysis Tool kit)进行变异检测及初步分析,其中使用到多种人类基因突变和疾病相关数据库,包括OMIM、1000G 和dbsnp 等。根据美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)的序列变异解释标准和指南[3],变异的致病性分为5 级:致病的(pathogenic,P),可能致病的(likely pathogenic,LP),意义不明确的(variant of uncertain significance,VUS),可能良性的(likely benign,LB)和良性的(benign,B)。一般情况下FLNC截断变异被归类为致病的(P);除非有来自共分离或功能性实验的额外证据,错义变体被归类为意义不明确的(VUS)。使 用 Polyphen-2、SIFT、PROVEAN 和 Mutation Taster 预测错义变异,使用PROVEAN 和Mutation Taster 预测无义变异、插入和缺失(Insertion and Deletion,InDel),筛选出被解读为致病的(P)和可能致病的(LP)变异,进行Sanger测序验证及临床研究。
致病性预测变异筛选及验证流程见图1。满足下列条件的变异认为是可能致病的(P):(1)在dbSNP 等人群数据库中未报道;(2)变异位于进化上高度保守位点;(3)在人类基因突变数据库中已有记录的心肌病及离子通道病相关基因上的特殊变异类型,如移码变异、终止获得/缺失变异、剪接位点变异;(4)在家系中该变异与疾病表型共分离;如果家系资料不适用于共分离,则致病性判断通过预测软件判断均为有害或可能有害的。
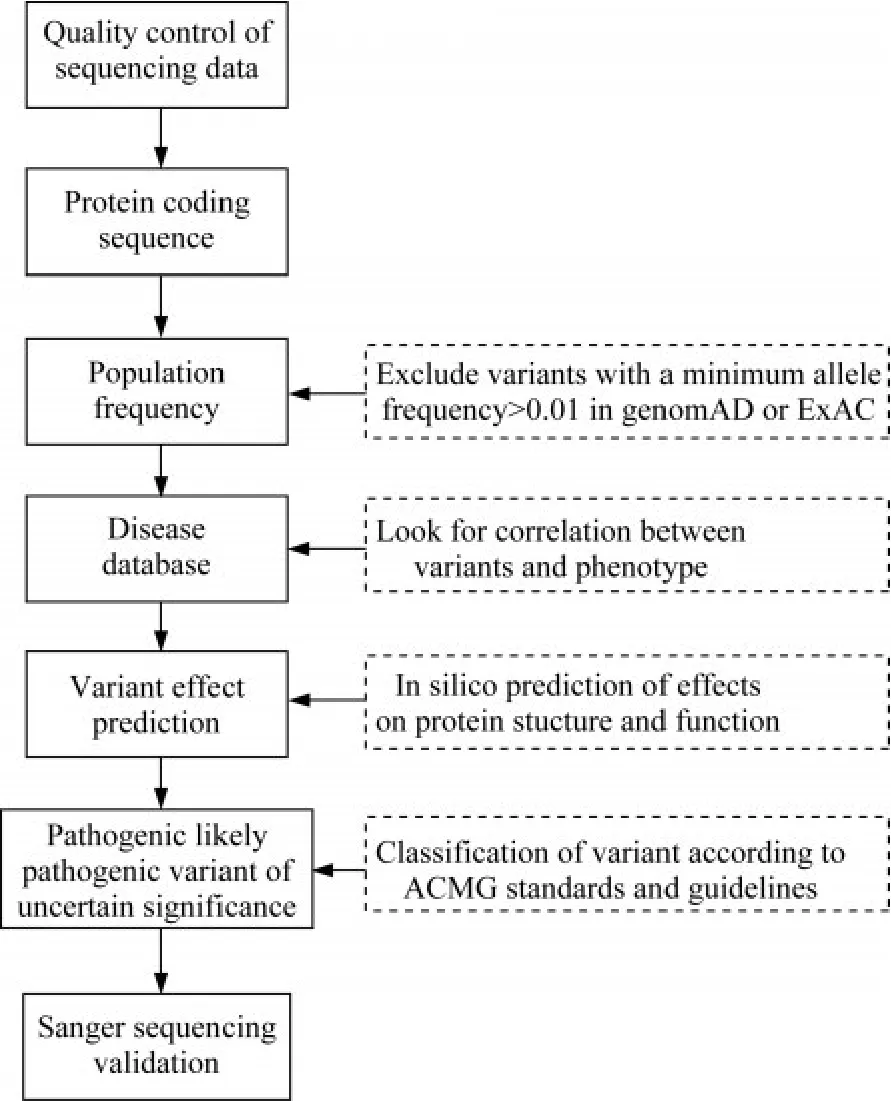
图1 患者全外显子测序及基因变异筛选流程图Fig 1 Flow chart of whole exome sequencing and variants screening
结 果
本研究共发现6 个FLNC变异(编号为1~6),均未见报道,包括HCM 患者的3 个错义变异,DCM 患者的1 个无义变异(截断变异)和1 个InDel变异,RCM 患者的 1 个错义变异,其中 1 例 DCM 为家族性,其余均为散发性病例。6 名患者均为男性,先证者及携带相同变异的家属详细临床资料见表1,各变异的位点、类型、致病性预测等数据见表2 及图 2。
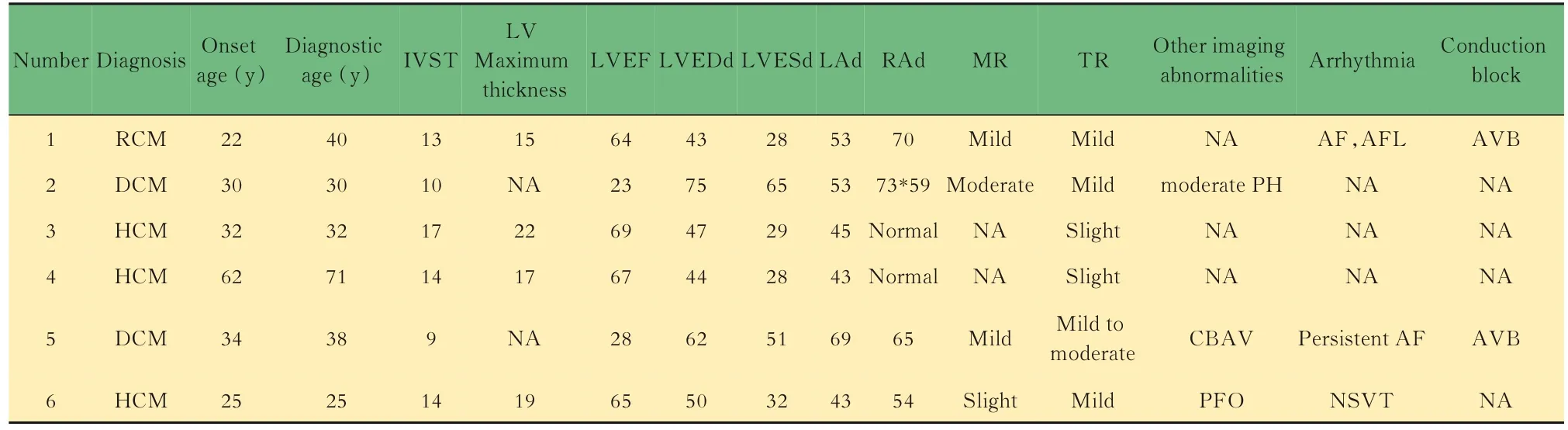
表1 携带FLNC 变异的心肌病患者的临床信息Tab 1 Clinical information of cardiomyopathy patients with FLNC variation

表2 患者携带的FLNC 变异信息Tab 2 Information of the FLNC variants detected in this study
1 号 RCM 患者,男性,44 岁,20 年前情绪激动后晕厥,在当地医院诊断为心肌肥厚,但未进一步随访和诊疗,直至4 年前频繁出现心悸、活动后气促,呈进行性加重,平地行走50 m 以上不能耐受,遂至当地医院检查,ECG 示心房扑动和心房颤动伴快速心室率。2017 年12 月患者就诊于我院,TTE 示:左室壁厚度13~22 mm,左室及右室收缩活动减弱,LVEF49%,双房明显增大伴轻度二尖瓣及三尖瓣反流;诊断为非梗阻性HCM,予抗凝、控制心室率等治疗,症状有所缓解。1 年前因频发心悸再次就诊,2019 年 7 月 ECG 示心房扑 动,2:1 房室传导;TTE 示:左右室壁增厚伴左右室壁整体收缩活动减弱(LVEF 50%),双房增大伴轻度二尖瓣及三尖瓣反流;心脏MRI 示:符合限制性心肌病改变,心肌淀粉样变待排;进一步行免疫固定电泳,结果呈阴性,最终确诊为RCM。
2 号DCM 先证者携带FLNC的截断变异及MYBPC3的错义变异(测序峰图及临床资料见图2),因患者母亲没有出现心肌病相关表型,未进行基因检测,无法证实是否为FLNC的新发变异;MYBPC3错义变异为罕见变异(GnomAD 中频率为0.000016),预测为可能致病或有害的。15 年前出现劳累后胸闷,诊断为DCM,予卡维地洛、培哚普利等药物治疗,病情平稳,2011 年5 月随访TTE示:左室显著增大,左室心尖部见丰富海绵状心肌成蜂窝状伴左室壁整体收缩活动减弱,LVEF33%,继续药物治疗,2012 年至2017 年多次随访TTE,LVEF 波动于30%~47%,ECG 提示Ⅱ度二型房室传导阻滞,2017 年11 月因上呼吸道感染后出现急性左心衰伴呼吸性代谢性碱中毒,经过抗感染、强心、利尿等治疗,病情仍进一步恶化,2018 年1 月因心衰、肝性脑病死亡。其父亲携带了相同的MYBPC3变异,11 年前因反复胸闷气促诊断为DCM,Ⅱ度二型房室传导阻滞(最长R-R 间期3.2 s)并行起搏器植入术。2012 年TTE 示:左房室扩大伴左室整体收缩活动减弱,LVEF31%,右房增大,中度三尖瓣反流;后多次随访TTE 心功能无明显恶化,病情平稳。

图2 2 号DCM 患者及其父亲的家系图、测序峰图及临床资料Fig 2 The family tree,chromatogram and clinical data of DCM 2 and his father
5 号DCM 患者20年前体检时发现二叶式主动脉瓣,18 年前出现活动后胸闷,于当地诊断为阵发性房颤,左房增大,未系统治疗。10 年前胸闷症状明显加重,就诊于我院,诊断为DCM。2010 年7 月,TTE示:先天性二叶纵裂式主动脉瓣畸形,轻度主动脉瓣反流,左室增大伴左室壁收缩活动减弱,LVEF35%,双房增大伴轻度二尖瓣及中度三尖瓣反流;ECG 示:持续性房颤;于我院行 CRT-D 术。2018 年 3 月,随访心超示:CRT 术后双房及左室扩大,左右室整体收缩活动减弱,LVEF28%,病情平稳。
剩余3 例HCM 患者发病年龄分别为32、62 和25岁,室间隔和左室最大厚度分别为17、14、14 mm 和22、17、19 mm,3 名患者均表现出典型的劳力性呼吸困难和胸痛,就医后诊断为HCM,3 人均无左室流出道梗阻,LVEF 正常。予美托洛尔治疗,病情均稳定。
讨 论
Z 盘对于心脏的重要性众所周知,其维持着心脏正常的机械活动,也参与多种生化途径相关信号的接收和传输,FLNC 通过与多个Z 盘蛋白相互作用,可以承受肌节上的强收缩力[4]。FLNC 蛋白由3个结构域组成(图3),位于氨基端的肌动蛋白结合结构域由2 个钙调蛋白同源结构域组成,连接24 个重复串联的免疫球蛋白样结构域,免疫球蛋白样结构域又由ROD1 和ROD2 两个铰链区及羧基端的二聚化结构域组成,FLNC 蛋白形成二聚体后才能与肌动蛋白结合。FLNC 蛋白不同结构域及不同类型的致病变异会导致不同的临床表型。ROD2 域是丝蛋白C 与Z 盘蛋白之间相互作用的重要区域,此结构域的错义突变会通过显性负性作用产生异常的蛋白聚体(包含无法正常二聚的FLNC 和一些Z 盘相关蛋白),蛋白聚体在细胞质中积累导致肌节的排列及功能紊乱,产生 HCM 和 RCM[5-6];位于肌动蛋白结合域的错义突变会产生毒性的功能获得,增强肌动蛋白结合活性导致肌病;而整个基因上的截断突变与DCM 和ACM 有密切关联,其通过无义介导的mRNA 降解机制产生基因单倍体不足,导致Z盘增厚紊乱,细胞间黏附减弱,影响横纹肌的机械传导[7-11]。携带FLNC错义突变的骨骼肌病患者有一小部分存在心脏表型[12],而FLNC相关心肌病患者罕有骨骼肌受累[7]。本研究中所有患者均无骨骼肌受累的表型,为什么FLNC错义突变相关HCM、RCM 患者骨骼肌中无蛋白聚体,现在还难以解释。修饰基因对FLNC表现度的影响、表观遗传学因素等可能共同作用于FLNC突变表型的复杂性,如果能够发现相同突变激活的不同通路,对于寻找潜在的治疗靶点可能存在重要的意义。
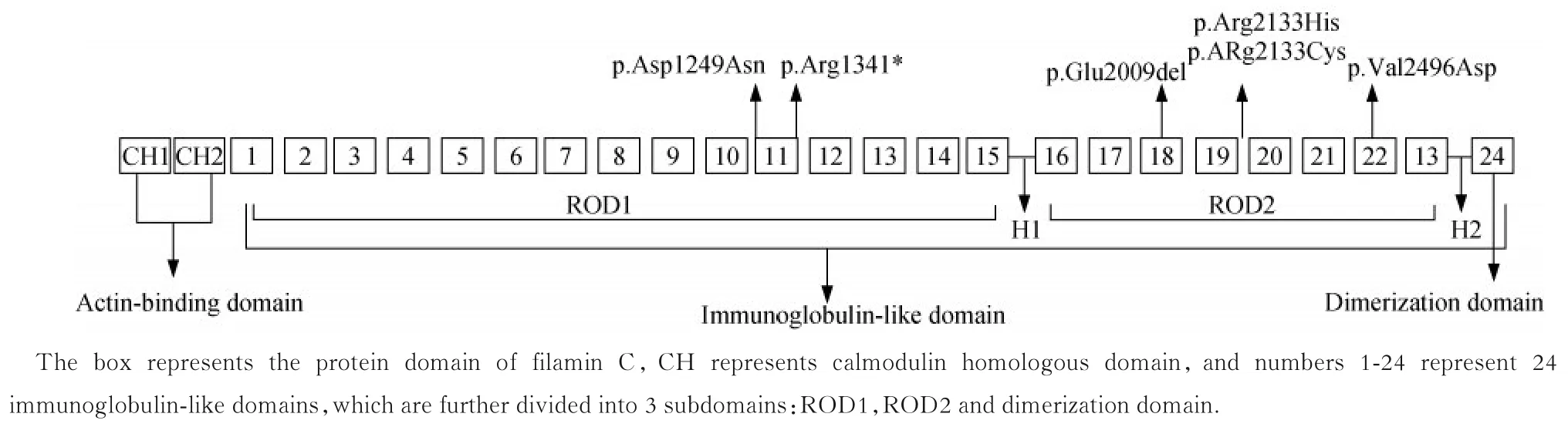
图3 FLNC 的结构域及本研究发现的变异分布Fig 3 Structure domains and variations distribution of FLNC in our research
以往研究证明FLNC变异在HCM 患者中较常见,其与肌节蛋白突变导致的HCM 在临床表型及预后方面没有明显差异[13],本研究中 3 个 HCM 患者携带的FLNC错义变异预测为可能致病,但是临床意义未知,因此FLNC变异对于HCM 患者的表型影响可能需要谨慎解释。在以往报道的病例中,FLNC相关DCM 的恶性程度更高,表现出与心室扩张程度不符的室性心律失常、心源性猝死高风险等特点[14]。本研究中 2 例 DCM 患者表现为发病年龄早,心功能恶化速度快,未观察到恶性室性心律失常;对于多种心肌病、多个基因突变或复合突变的携带者终末期进展、心律失常和预后不良的风险明显增加[15-17],2 号 DCM 患者同时携带双基因杂合变异,且均预测为可能致病的变异,与仅携带单基因变异的患者父亲相比,临床表现为发病年龄早,心功能恶化速度快伴随传导系统受累,这也符合之前的研究结果。有不同的研究者在HCM 及RCM患者中发现了相同的错义突变(p.Ser1624Leu),提出携带该突变的家系可能存在HCM 和RCM 混合的表型[12,18];本研究中,RCM 患者在发病初期仅表现为心肌肥厚,早期诊断为HCM,随着病程进展出现双房扩大,舒张功能明显受限,最终确诊为RCM,3 号HCM 患者与RCM 患者携带的变异碱基位点相邻,导致相同的氨基酸替换,随着年龄增加是否会进展成为RCM 尚不明确,我们将对此患者进行定期随访。
本研究对200 名汉族不同亚型心肌病患者进行全外显子组测序,讨论了在200 例汉族心肌病患者中FLNC基因的变异特点,扩大了心肌病中FLNC基因的变异谱,并进一步探讨了在不同心肌病中FLNC变异的基因型和表型相关性。我们观察到变异类型对于临床特点有决定性作用,错义突变干扰了蛋白质的二聚和折叠会导致HCM、RCM,截断突变会通过单倍体不足导致细胞间黏附减弱,产生发病年龄早、进展快速的DCM。本研究不足之处在于作为单中心研究纳入的人数较少,且未纳入ACM 患者,这种心肌病中FLNC的突变谱及机制需要进一步的挖掘,另外本研究发现的变异需要在更大样本的心肌病患者中进行对比和验证,同时获得更多功能验证实验的证据,以进一步确定其致病性。
作者贡献声明李雪洁 数据采集,论文撰写。周年伟,孙敏敏,李伟,汪咏莳,谢惠琳 数据采集。舒先红 论文修订。
利益冲突声明所有作者均声明不存在利益冲突。
