高效液相色谱法同时测定食品中两种非食用色素的含量
2021-10-12◎李涛,周琼
◎ 李 涛,周 琼
(1安康市质量技术检验检测中心,陕西 安康 725000;2陕西省富硒食品工程实验室,陕西 安康 725000)
近年来,一些不法食品经营者为了获取更大利益,打着食品添加剂的旗号,在食品中添加非食用物质,对人们的生命和健康造成巨大伤害,如三聚氰胺、吊白块、苏丹红、碱性橙等,引发了重大的食品安全事故。
在这些非法添加物中,苏丹红、碱性橙这两种非食用色素被使用的频率较高。苏丹红和碱性橙种类多样,单独或重叠使用都能使产品呈现良好的色泽,且成本低廉,增加了产品竞争力。国家技术部门出台了相应的检测方法标准[1-2],只能对苏丹红和碱性橙分别进行检测,检测效率较低。若能同时检测这两种非食用色素,则能很大程度地提高检测效率、降低检验成本。本研究结合苏丹红和碱性橙的检测条件,建立了同时检测苏丹红和碱性橙的分析方法,快速准确,具有一定的参考价值。
1 材料与方法
1.1 材料与试剂
豆腐干、豆油皮、腐竹、豆腐乳、豆瓣酱、辣椒酱和辣椒面等,购自陕西省安康市本地市场;标准品[碱性橙2(CAS号:532-82-1,纯度≥99.9%)、碱性橙21(CAS号:3056-93-7,纯度≥96.7%)、碱性橙22(CAS号:4657-00-5,纯度≥99.9%)],购自海安谱实验科技股份有限公司;苏丹红Ⅰ(编号:GBW10056纯度为≥99.9%),购自中国计量科学院研究院;所有试验用水符合GB/T 6682—2008中一级水要求(超纯水);乙酸铵(分析纯);无水硫酸钠(分析纯);氨水(分析纯);甲醇(色谱纯);乙腈(色谱纯);磷酸(分析纯)。
1.2 仪器与设备
高效液相色谱仪:LC-20A,岛津公司;紫外检测器;C18色谱柱(岛津Inertsil公司250 mm×4.6 mm,5 μm);超声波清洗仪;超纯水器;电子天平;离心机;针头过滤器:孔径为0.45 μm有机微孔滤膜。
1.3 样品处理
将样品搅碎,称取2 g(精确至0.001 g)样品于25 mL离心管中,加入1 mL蒸馏水润湿样品;用10%(V/V)氨水调节样品至pH为7~8,加入5 mL乙腈提取,振荡10 min,超声10 min,4 000 r·min-1离心5 min;将上清液移入10 mL容量瓶中,向残渣中再加入5 mL碱性甲醇,振荡10 min,超声10 min,4 000 r·min-1离心5 min;合并上清液,用甲醇定容至10 mL;过0.45 μm滤膜,供HPLC分析[3-5]。
1.4 标准曲线绘制
1.4.1 标准溶液制备
分别称取标准品碱性橙2、碱性橙21、碱性橙22、苏丹红Ⅰ各50 mg,(精确至0.000 1 g),置于100 mL棕色容量瓶中,加90%乙腈超声使之完全溶解,定容,摇匀。所配溶液中碱性橙2、碱性橙21、碱性橙22、苏丹红Ⅰ浓度均为500 μg·mL-1标准储备液,于4 ℃保存。
1.4.2 标准中间液
准确移取碱性橙2、碱性橙21、碱性橙22、苏丹红Ⅰ标准贮备液各5 mL,分别置于50 mL棕色容量瓶中,用90%乙腈定容至刻度。所配溶液中碱性橙2、碱性橙21、碱性橙22、苏丹红Ⅰ浓度均为50 μg·mL-1,于4 ℃保存。
1.4.3 标准工作溶液
准确移取碱性橙2、碱性橙21、碱性橙22、苏丹红Ⅰ染料标准中间液各0.5 mL、1.0 mL、2.0 mL、5.0 mL、7.5 mL和10.0 mL于50 mL棕色容量瓶中,用90%乙腈定容至刻度。此时,混合溶液中碱性橙2、碱性橙21、碱性橙22、苏丹红Ⅰ染料的浓度分别为0.5 μg·mL-1、1.0 μg·mL-1、2.0 μg·mL-1、5.0 μg·mL-1、7.5 μg·mL-1和10.0 μg·mL-1。将配制好的标准工作溶液,绘制以峰面积为纵坐标,工作溶液浓度为横坐标的标准曲线。
1.5 色谱条件
色 谱 柱:C18(250 mm×4.6 mm,5 μm),岛津Inertsil公司;流动相:甲醇-乙酸铵(pH 4.5,20 mmol·L-1);流速以1.0 mL·min-1进行梯度洗脱,梯度洗脱程序见表1;柱温:40 ℃;检测器:紫外检测器;检测波长:450 nm;进样量:20 μL。
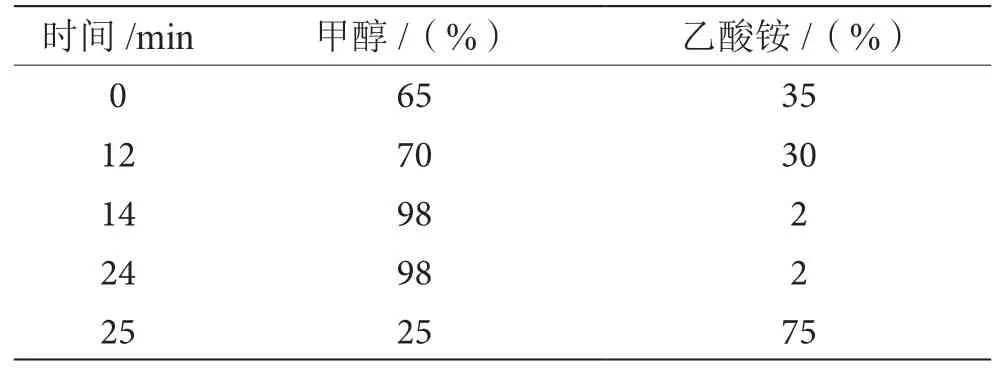
表1 梯度洗脱程序表
1.6 样品分析
按照1.5中的色谱条件检测待测样品,绘制标准曲线并比较,依据保留时间进行定性分析,采用外标峰面积法进行定量分析。
2 结果与分析
2.1 色谱条件选择
2.1.1 流动相的选择
根据苏丹红和碱性橙的分析方法,采用梯度洗脱程序,分别试验了甲醇-水、甲醇-乙腈-水、甲醇-乙酸铵溶液等作为流动相,探究其对碱性橙和苏丹红分离效果的影响,结果发现,甲醇-乙酸铵作为流动相时,苏丹红和碱性橙的分离效果、峰型及线性关系都比较理想,采用其他流动相时,或有组分缺失,或有几种组分重叠无法分离的问题。
关于流动相中乙酸铵的浓度,分别采用20 mmol·L-1和50 mmol·L-1乙酸铵进行试验,二者都能达到良好效果,且50 mmol·L-1时响应值稍高,但柱压相应增加,在分离效果方面二者无大的差别。若乙酸铵浓度高,溶质在系统中析出程度增加,对系统的稳定和后期冲洗造成影响。因此,为避免高浓度乙酸铵在系统中析出的影响,本试验采用20 mmol·L-1乙酸铵作为流动相。同时参考有关资料,对乙酸铵在不同pH值下的分离效果进行比较试验[3],用1 mol·L-1磷酸调节pH值为3.5、4.5、5.5和6.5,结果发现当pH值为4.5时,效果最好。但结合实际,不同的色谱柱性能有差异,对酸度的适应有所不同,不一定每支色谱柱在pH值为4.5时效果最好。
关于梯度洗脱条件,上述条件只是其中一种。甲醇含量增加,分离速度快,但分离效果降低;甲醇含量降低,分离速度减慢,分离效果变好。如果降低初始甲醇含量至30%左右,减缓甲醇比例上升速度,则分离效果更好一些,但4种组分出峰时间会相应延长2~5 min。
2.1.2 检测波长的选择
有关资料显示[2],碱性橙2在波长450 nm处有最大吸收,碱性橙21、碱性橙22和苏丹红Ⅰ在480 nm处有最大吸收,本试验对这两个波长进行了比较,结果发现,在波长为450 nm时,4种组分都能达到满意的检测效果,而在480 nm时,碱性橙2的响应值不太理想,故而选择波长为450 nm。
2.2 前处理条件选择
提取碱性橙的溶剂有甲醇和乙醇,提取苏丹红的溶剂有乙腈和丙酮等,单独使用某一种或某一类,都不能将几种组分完全提取出来,回收率很低。经过试验发现,乙腈提取苏丹红效果较好,碱性甲醇提取碱性橙效果较好,乙醇提取峰形杂乱不利于定性,故而采取乙腈-碱性甲醇的提取组合。调节pH值是为了使各个样品保持统一稳定的酸碱环境,保证方法的重现性和稳定性,保证被检组分的定性检出。
2.3 标准曲线和检出限
用1.5中的色谱条件分析4种非法添加色素(浓度均为10.0 mg·kg-1),标样谱图如图1所示。4种组分得到很好分离。在标准系列中,4种组分的浓度分别为1.0 mg·L-1、2.0 mg·L-1、5.0 mg·L-1、7.5 mg·L-1和10.0 mg·L-1,在此范围内,峰面积与浓度之间有良好的线性关系。同时测定4种组分的检出限,均达到0.05 mg·L-1,具体如表2所示。

图1 4种非法色素检测谱图

表2 4种组分线性范围、回归方程、线性相关系数和检出限表
2.4 回收率和精密度试验
分别选取2.0 g辣椒面和豆腐乳样品(均未含碱性橙和苏丹红),添加4种组分的混合标准溶液,每个组分添加高低浓度各两个平行样,进行加标回收试验。回收率、相对相差和定量限如表3所示。结果表明,两种样品辣椒面和豆腐乳的两平行试验中碱性橙的回收率均大于80%,苏丹红Ⅰ的回收率≥89%。相对相差在3.8%~4.6%,4种组分的定量限在0.20 mg·kg-1。
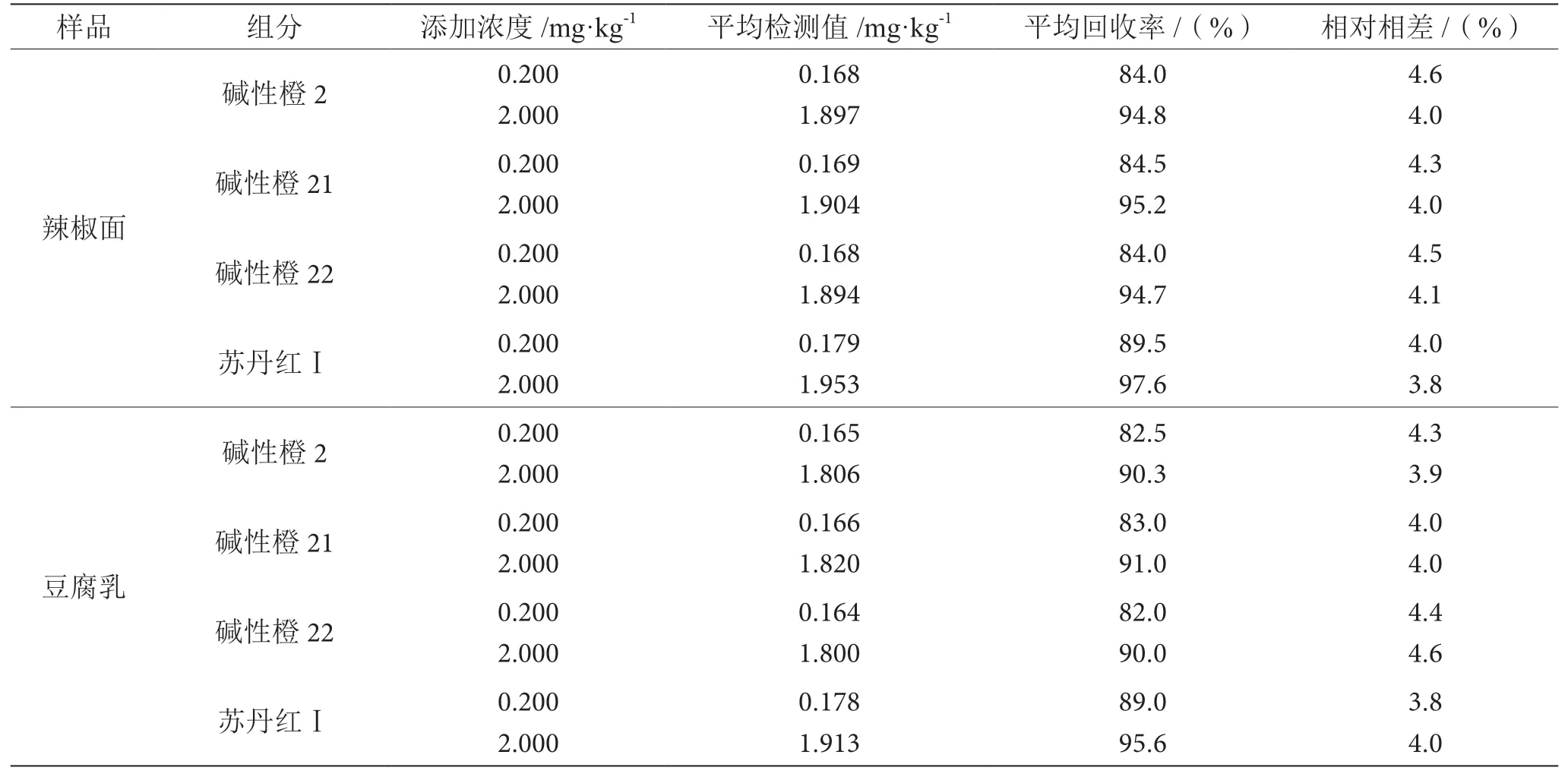
表3 4种组的加标回收率、相对偏差表
2.5 样品的含量测定
采用本方法对市售的豆腐干、豆油皮、腐竹、豆腐乳、豆瓣酱、辣椒酱和辣椒面共20份样品中的非食用色素进行了检测,具体情况见表4。

表4 不同食品中的色素检测结果表
3 结语
采用本方法对安康本地产的辣椒面、辣椒酱、豆腐乳、豆瓣酱、腐竹、豆油皮等产品进行检测,均未检测出碱性橙和苏丹红。而且安康当地也没有非法添加苏丹红和碱性橙的投诉,也没有其他检测机构在安康出产的食品中检测出碱性橙和苏丹红的报告。本方法稳定可靠,快速准确,重现性好,检测效率高,具有一定的参考价值。
