同位素内标-超高效液相色谱串联质谱法测定饮料中咖啡因的含量
2021-10-12康雪梅郭倩倩李梦雨贾松涛赵雪峰赵林萍
◎ 康雪梅,陶 燕,郭倩倩,李 新,李梦雨,贾松涛,赵雪峰,赵林萍
(河南中标检测服务有限公司,河南 郑州 450001)
咖啡因是一种黄嘌呤生物碱化合物,是一种中枢神经兴奋剂,能够暂时驱走睡意并恢复精力,临床上用于治疗神经衰弱和昏迷复苏。有咖啡因成分的咖啡、茶、软饮料及能量饮料十分畅销,因此,咖啡因也是世界上最普遍被使用的精神药品。过多的饮用摄入大量的咖啡因,导致作息不规律引发精神紊乱;还会引起肠痉挛。长期饮用会引发慢性胃炎,刺激肾功能,导致儿童多尿,流失大量钙质,影响骨骼发育。2017年10月27日,世界卫生组织国际癌症研究机构公布的致癌物清单初步整理参考,咖啡因属于3类致癌物,即“尚不能分类”的致癌物[1]。
目前,咖啡因检测方法主要有碘量法、HPLC法、近红外光谱分析法、毛细管电泳法等。近几年对于液质法的使用逐渐增多,但基本也是外标法的建立使用[2-7]。外标法对于复杂基质的测定难于避免基质效应等部分的影响。因此本文旨在建立同位素内标-超高效液相色谱串联质谱法测定饮料中咖啡因含量的方法。
1 材料与方法
1.1 试验材料
碳酸饮料(可乐)、功能型饮料(红牛)、绿茶、咖啡等。
1.2 试剂及仪器设备
甲醇(色谱纯,美国赛默飞);乙腈(色谱纯,美国赛默飞);甲酸(色谱纯,美国赛默飞);咖啡因对照品(批号G138494,纯度98.8%,美国DR);咖啡因-D3同位素内标对照品(批号449472,100 mg·L-1,o2si);试验用水为去离子水。
TSQ vanquish超高效液相色谱-三重四级杆串联质谱仪(美国赛默飞世尔科技);天平,感量为0.1 mg(美国奥豪斯);水浴锅;超声波清洗器,0.45 µm微孔水相滤膜(津腾)。
1.3 试验方法
1.3.1 对照品溶液的制备
(1)咖啡因标准储备液(1 mg·mL-1)。准确称取咖啡因标准品10 mg(精确至0.01 mg)于10 mL容量瓶中,用甲醇溶解并定容,4 ℃冰箱保存。
(2)咖啡因标准中间工作液(1 µg·mL-1)。准确吸取0.1 mL咖啡因标准储备液于100 mL容量瓶中用水定容后,4 ℃冰箱保存。
(3)咖啡因-D3同位素内标标准中间工作液(1 µg·mL-1)。准确吸取1 mL咖啡因标准储备液于100 mL容量瓶中用水定容后,4 ℃冰箱保存。
(4)咖啡因标准曲线工作液。分别吸取咖啡因标准中间液0.05 mL、0.10 mL、0.20 mL、0.50 mL、1.00 mL和2.00 mL于10 mL容量瓶中,向其中分别加入0.1 mL咖啡因-D3同位素内标标准中间工作液,用水定容,得到咖啡因浓度分别为5.0 ng·mL-1、10.0 ng·mL-1、20.0 ng·mL-1、50.0 ng·mL-1、100.0 ng·mL-1和200.0 ng·mL-1,内标浓度为10 ng·mL-1,临用时配制。
1.3.2 样品的制备净化过程
(1)可乐型饮料。①脱气。样品用超声清洗器在40 ℃下超声5 min。②净化。称取5 g(精确至0.001 g)样品,加入咖啡因内标标准中间工作液(1 µg·mL-1)100 µL,加水定容至100 mL,摇匀,加入0.5 g氧化镁,振摇,静置,取上清液经微孔滤膜过滤,备用。
(2)不含乳的咖啡及茶叶液体制品。称取5 g(精确至0.001 g)样品,加水定容至100 mL,摇匀,加入0.5 g氧化镁,振摇,静置,取上清液经微孔滤膜过滤,备用。
(3)含乳的咖啡及茶叶液体制品。称取1 g(精确至0.001 g)样品,加入三氯乙酸溶液(10 g·L-1)定容至100 mL,摇匀,静置,沉降蛋白,取上清液经微孔滤膜过滤,备用。
若样品溶液中咖啡因含量超出标准曲线范围需再做稀释,相应内标加入量要增加,使内标上机浓度为10.0ng·mL-1。
1.3.3 仪器条件
(1)色 谱 条 件。色 谱 柱,waters ACQUITY UPLC BEH C18,粒径1.7 µm,2.1 mm×100 mm;流动相:0.1%甲醇+0.1%甲酸水;流速:0.25 mL·min-1;柱温:25 ℃;进样量,2 µL。
(2)质谱条件。离子源温度:350 ℃;离子化方式:ESI+源;多反应监测模式。
1.3.4 标准曲线的制作
将标准系列工作液分别注入液相色谱仪中,测定相应的峰面积,以标准工作液的浓度为横坐标,以咖啡因和咖啡因-D3峰面积比值为纵坐标,绘制标准曲线。
1.3.5 试样溶液的测定
将试样溶液注入液相色谱仪中,以保留时间定性,以保留时间及离子峰度比双重定性,以咖啡因和咖啡因-D3峰面积比值带入标准曲线定量。
2 结果与分析
2.1 质谱条件的选择及优化
采用不经液相,直接手动和自动注射的方式,在正、负模式下切换,选择合适的电离模式,结果显示咖啡因在正离子模式下拥有较好的响应度。然后在正离子模式下对咖啡因及咖啡因-D3进行Q1母离子及Q3子离子进行优化,选取1个母离子和2个最优子离子,并分别得到碰撞能,确定定性离子对和定量离子对,结果见表1。咖啡因及咖啡因D3的总离子流图和提取离子色谱图、棒状图见图1、图2和图3。

图1 饮料中咖啡因和咖啡因-D3经LC/MSMS分析得到的总离子流图
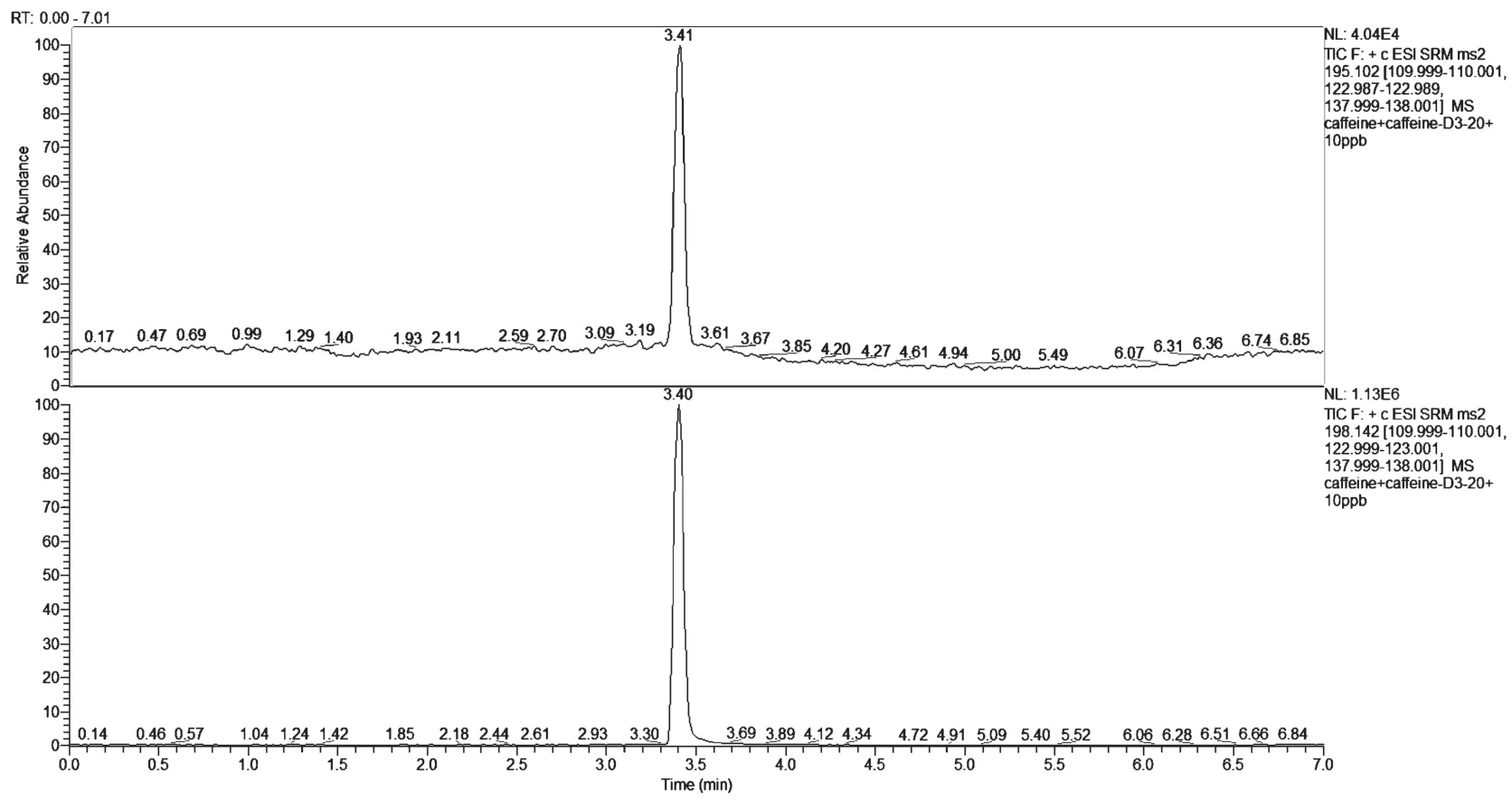
图2 饮料中咖啡因和咖啡因-D3经LC/MSMS分析得到的提取离子色谱图
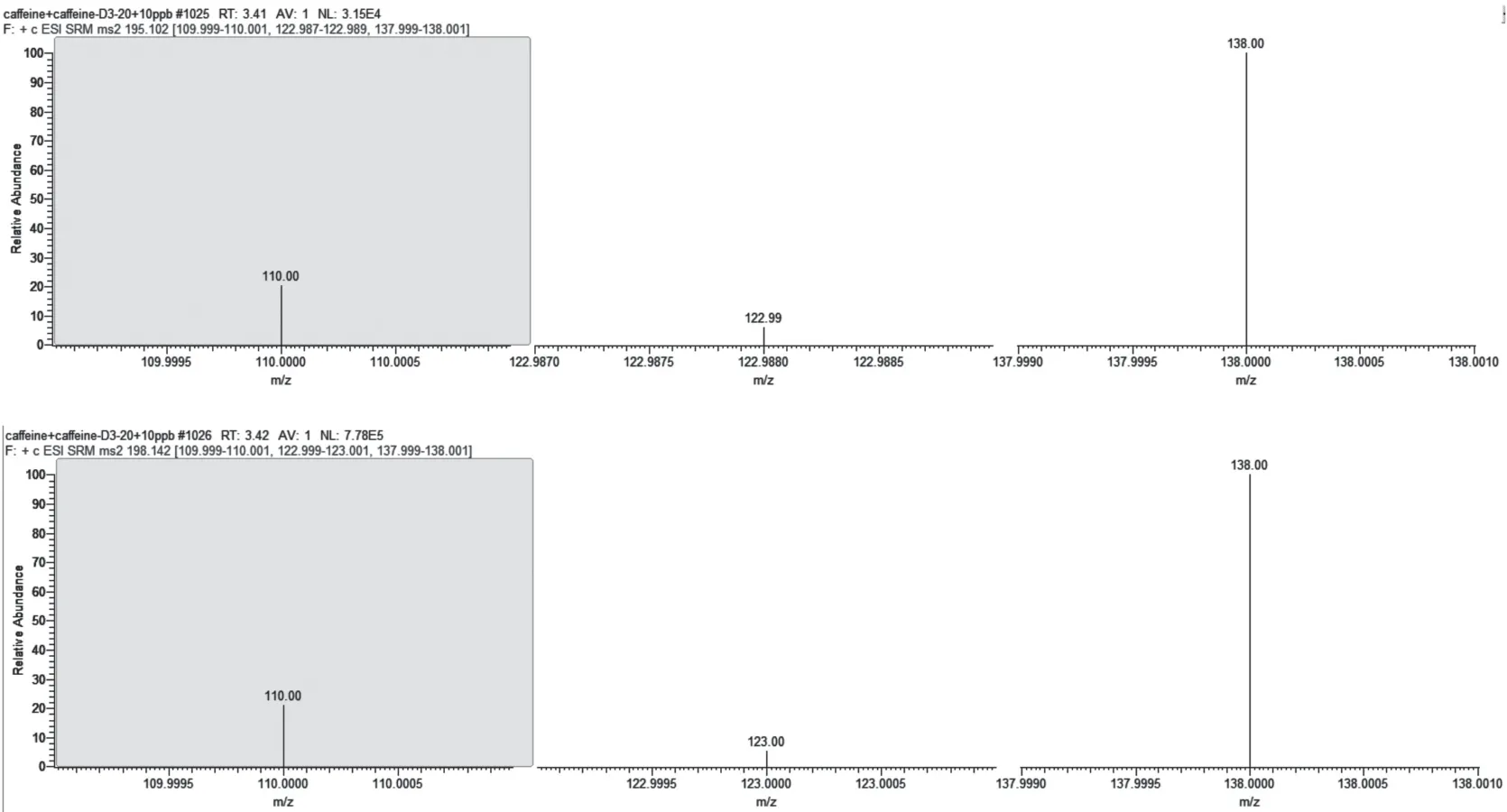
图3 饮料中咖啡因和咖啡因-D3经LC/MSMS分析得到的提取离子棒状图

表1 咖啡因和咖啡因-D3的质谱多反应监测检测条件表
2.2 线性范围、检出限与定量限研究
用试剂分别配制浓度为0.1 ng·mL-1、 0.2 ng·mL-1、0.5 ng·mL-1、1.0 ng·mL-1L、2.0 ng·mL-1、5.0 ng·mL-1、10.0 ng·mL-1、20.0 ng·mL-1、50.0 ng·mL-1、100.0 ng·mL-1、200.0 ng·mL-1、500.0 ng·mL-1、1 000.0 ng·mL-1、2 000.0 ng·mL-1的咖啡因标准溶液,在上述质谱条件下测定,以浓度和峰面积绘制标准曲线。结果表明,在0.2~500.0 ng·mL-1浓度范围内,饮料中咖啡因有较高的线性拟合度,以SN>3时标线的浓度对应样品的浓度为检出限,以SN>10时标线的浓度对应样品的浓度为定量限。本法咖啡因的检出限为0.2 ng·mL-1(相当于1 g样品定容体积为100 mL时,检出限为20 ng·mL-1);定量限为0.5 ng·mL-1(相当于1 g样品定容体积为100 mL时,定量限为50 ng·mL-1)。
2.3 回收率、方法重复性研究
由于市售绿茶、红牛、咖啡等饮料咖啡因含量不均,且浓度较高,添加低浓度的咖啡因研究其回收率重复性无法获取准确结果,因此本试验将咖啡因-D3标准物质添加到可乐、绿茶、红牛、咖啡中,添加浓度水平为20 ng·mL-1、50 ng·mL-1、200 ng·mL-1的质控浓度溶液,平行试验10次,计算加标回收率和精密度,结果见表2。由表2可知,待测物的回收率均大于84.3%,精密度均小于12.0%。
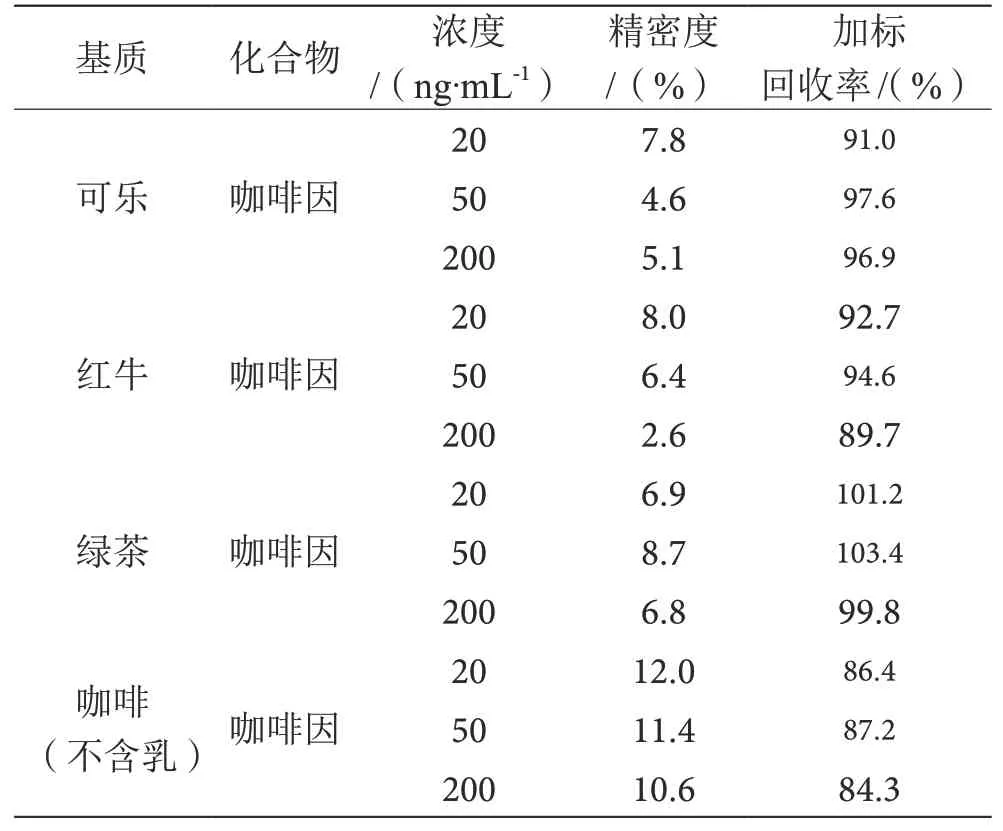
表2 方法的精密度和加标回收率表(n=10)
2.4 实际样品的检测
采用本方法对市售的红茶、绿茶、咖啡、可乐、茶饮料等20个市售饮品进行测定。结果显示,咖啡因均有检出,含量在50~2 000 µg·mL-1,这说明市售的未标明咖啡因含量的饮料也或多或少含有一定量的咖啡因,对于儿童等不易食用咖啡因的人群来说是潜在的隐患。
3 结论
本试验基于LC-MS/MS同位素内标法建立了市售饮料中咖啡因的快速定性定量分析方法。该方法简单易行,分析时间短,灵敏度高,重复型好,不仅能够满足日常实际检测要求,对于痕量样品的分析同样适用。
