Lipid droplets as metabolic determinants for stemness and chemoresistance in cancer
2021-10-11AlbaRoyoGarcSarahCourtoisBeatrizParejoAlonsoPilarEspiauRomeraPatriciaSancho
Alba Royo-García, Sarah Courtois, Beatriz Parejo-Alonso, Pilar Espiau-Romera, Patricia Sancho
Alba Royo-García, Sarah Courtois, Beatriz Parejo-Alonso, Pilar Espiau-Romera, Patricia Sancho, Hospital Universitario Miguel Servet, IIS Aragón, Zaragoza 50009, Spain
Abstract Previously regarded as simple fat storage particles, new evidence suggests that lipid droplets (LDs) are dynamic and functional organelles involved in key cellular processes such as membrane biosynthesis, lipid metabolism, cell signalling and inflammation. Indeed, an increased LD content is one of the most apparent features resulting from lipid metabolism reprogramming necessary to support the basic functions of cancer cells. LDs have been associated to different cellular processes involved in cancer progression and aggressiveness, such as tumorigenicity, invasion and metastasis, as well as chemoresistance. Interestingly, all of these processes are controlled by a subpopulation of highly aggressive tumoral cells named cancer stem cells (CSCs), suggesting that LDs may be fundamental elements for stemness in cancer. Considering the key role of CSCs on chemoresistance and disease relapse, main factors of therapy failure, the design of novel therapeutic approaches targeting these cells may be the only chance for long-term survival in cancer patients. In this sense, their biology and functional properties render LDs excellent candidates for target discovery and design of combined therapeutic strategies. In this review, we summarise the current knowledge identifying LDs and CSCs as main contributors to cancer aggressiveness, metastasis and chemoresistance.
Key Words: Lipids; Lipid droplets; Lipid metabolism; Stemness; Cancer stem cells; Chemoresistance
INTRODUCTION
Cancer stem cells
The cancer stem cell concept: Consistent evidence supports that most of the heterogeneity found in both liquid and solid cancers might be originated in the context of hierarchical organisation of the tumours. Indeed, a subset of cells with self-renewal capacity and tumour-initiating properties called cancer stem cells (CSCs) undergo asymmetrical and symmetrical divisions in order to originate bulk differentiated tumour cells and identical CSCs to perpetuate its lineage. Cancer hierarchy at cellular level was first described in acute myeloid leukaemia[1], representing a huge milestone in the understanding of cancer emergence. The CSC theory has been supported since then by increasing evidence in other malignancies such as breast cancer[2], brain tumours[3] colon and colorectal cancers[4,5], as well as pancreatic cancer[6,7], among others.
The current approach to cancer therapy has been both clarified and challenged by the existence of CSCs. On the one hand, the increasing evidence of their existence and contribution to tumorigenesis and metastasis has allowed researchers and clinicians to acquire a better understanding of cancer origin and evolution. On the other hand, proof of the implication of CSCs in treatment failure due to their intrinsic chemoresistance abilities has demonstrated that specific therapeutic strategies against this tumoral subpopulation are still urgently needed.
The origin of CSCs remains unclear, since it might vary between malignancies. One hypothesis derives from the observed similarities between CSCs and their normal homologous SCs, suggesting that local SCs may suffer a malignant transformation[8]. Other theories involve the acquisition of stemness features by differentiated cells. On the one hand, it has been suggested that differentiated cancer cells undergoing epithelial-to-mesenchymal transition acquire stem-like properties under the regulation of Notch signalling[9,10]. On the other hand, microenvironmental signals from stromal cells might facilitate non-CSCs dedifferentiation. For instance, Wnt signalling conferred self-renewal and tumorigenic abilities to colorectal cancer cells[11]. Furthermore, FGF5 and collagen production induced by Hedgehog promoted triple negative breast cancer chemoresistance by acquiring self-renewal capacity[12]. In any case, a dual scenario in which both local SCs and differentiated tumour cells originate new CSCs may be present in chemoresistant pancreatic[13] and lung[14] cancer cells.
CSC metabolism
Microenvironmental selective pressure forces CSCs to adapt continuously in order to survive and progress. For instance, as the tumour grows, glucose and oxygen levels diminish, the pH becomes acidic and reactive oxygen species (ROS) and inflammatory mediators accumulate in the tumour microenvironment. Since most differentiated tumour cells are fully glycolytic in order to cope with their enhanced proliferative rates (e.g.Warburg effect), resource scarcity forces CSCs to become metabolically and functionally plastic in order to survive and detoxify their microenvironment. Theoretically, an active mitochondrial metabolism would provide CSCs with an increased plasticity since a larger array of substrates could be feeding the tricarboxylic acid cycle. However, depending on the tumour type and model systems studied, CSCs use either mitochondrial oxidative phosphorylation (OXPHOS) or glycolysis[15,16] preferentially, with varying degrees of plasticity to switch from, even within the same tumour. Indeed, although the majority of pancreatic CSCs relies on OXPHOS and is very sensitive to mitochondrial inhibition, a small portion of CSCs shows a plastic phenotype, activating glycolysis when its mitochondria are inhibited[17]. However, full metabolic plasticity comes at the expense of self-renewal capacity[17].
Importantly, OXPHOS-dependent CSCs and therapy-resistant tumour cells from different cancer types bear higher levels of the master regulator of mitochondrial biogenesis peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α)[17-19], which supports OXPHOS metabolism and provides resistance to oxidative stress and chemotherapy[18-20]. Considering that PGC-1α is a transcriptional coactivator of the peroxisome proliferator-activated superfamily of receptors (PPARs) which controls the balance between glucose and lipid metabolism[21,22], we can hypothesise that PGC-1α enables CSCs to control a complex metabolic programme associating stemness to mitochondrial metabolism, including lipid and fatty acid (FA) oxidation (FAO). In fact, different studies have demonstrated that lipid metabolism is required to maintain the CSC pools in several tumour types[23-26].
LIPID METABOLISM AND LIPID DROPLETS
Cancer and lipid metabolism
Cancer cells have metabolic reprogramming abilities to sustain high proliferation rates as well as energy production, not only through high glycolysis (Warburg effect), but also through reprogrammed lipid metabolism[27-29]. Indeed, they enhancede novolipid synthesis, lipogenesis and FAO, being FA synthesis one of the most important aberrations of cancer cell metabolism[30]. FAs are involved in many different aspects of tumorigenesis and tumour progression and sustain three requirements of cancer cells and CSCs: Cell membrane formation, signalling molecules and lipid-derived messengers, and energy production[31-33]. Importantly, an increased FA metabolism has been associated to poor prognosis in different types of cancer, such as pancreatic cancer or melanoma[34]. In pancreatic cancer, it is generally associated to a high expression of key regulatory enzymes like the FA synthase and sterol regulatory element-binding protein[35,36].
Cancer cells accumulate more lipids in their cytoplasm than normal cells[37]. Novikoff was the first to demonstrate the presence of cytoplasmic inclusions in the rat liver tumour cells and to identify the lipid nature of these droplets[38]. Although regarded as simple fat storage particles for long, lipid droplets (LDs) are currently considered conserved, dynamic and functional organelles involved in membrane biosynthesis, lipid metabolism, cell signalling and inflammation[39]. Indeed, they have been associated with an increased tumour aggressiveness and resistance to chemotherapy[40], considerably raising attention within the cancer biology community.
LDs
LDs, also known as lipid bodies or liposomes, are cellular organelles ranging from 20-40 nm to 100 mm, with key functions for lipid and energy homeostasis[41,42]. The quantity, size, composition and intracellular localisation differ significantly between or within cells, mainly due to their type, function and metabolic state[43]. Indeed, LDs are highly dynamic organelles which alternate periods of growth and consumption, depending on cell energy and nutritional status[39,41].
However, all LDs have a similar structure consisting of a hydrophobic core of neutral lipids, such as cholesteryl esters (CE), retinyl esters and triglycerides (TAGs)[44], separated from the aqueous cytoplasm by a monolayer of phospholipids, mainly phosphatidylcholine[45]. Additionally, LDs are coated with integral and peripheral proteins[46] derived from the cytosol or the endoplasmic reticulum (ER)[47]. These proteins can be classified into four groups: (1) Resident/structural proteins, such as members of the perilipin (PLIN)-ADRP-TIP47 family or the cell death-inducing DFF45-like effector (CIDE) family[48-50] (Figure 1); (2) Lipid metabolism enzymes, such as diacylglycerol acyltransferases 1 and 2 (DGAT1 and DGAT2), adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL); (3) Membrane trafficking proteins, including a variety of Ras related protein (Rab) GTPases, as well as soluble NSF binding protein receptor proteins; and (4) Cell signalling proteins such as mitogen-activated protein kinases and protein kinase C. Other types of proteins can be associated to the ribosome and cytoskeleton, or processes such as protein degradation[51,52].
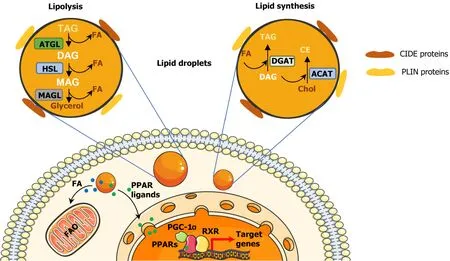
Figure 1 Structure and cellular functions of lipid droplets.
LD biogenesis can be described as an evolutionary model consisting of three main steps: (1) Lipid synthesis; (2) LD formation; and (3) LD growth. In step 1, TAG and CE synthesis enzymes, such as DGAT1, DGAT2 and acyl-CoA cholesterol acyltransferases 1 and 2 (ACAT1 and ACAT2), deposit neutral lipids between the sheets of the ER bilayer[53,54]. During step 2, the lipid quantity increases and, when it reaches a certain concentration, the LD detaches from the ER[55]. Thereafter, a variety of proteins such as perilipins, are recruited to the lens structure and facilitate the growth of the nascent LD[56]. Finally, step 3 only occurs in some mammalian cells, where LDs can grow by local lipid synthesis, by transporting lipids to LDs or by fusing with other LDs[57].
LDs can be broken down for energy supply and membrane synthesis through lipolysis or lipophagy (Figure 1). The lipolysis enables the release of FAs from TAGs through the consecutive action of ATGL, HSL and monoacylglycerol lipase[58,59]. Through lipophagy, LDs are enclosed in autophagosomes, fused with lysosomes and degraded by hydrolytic enzymes[60,61].
LDs are mainly found in the cytoplasm, but also in the nucleus of some cell types[62]. Their intracellular location is determined by interacting with other organelles to promote lipid exchange, metabolic dynamics and stress adaptation[63]. LDs come into contact with the ER early in their biogenesis, as well as with the lysosome in the lipophagy process[56,61]. LDs also connect with mitochondria to enable the direct flow of FAs into the mitochondrial matrix to fulfil the cell energy requirements[64]. Their interaction with peroxisomes also allows the transport of FAs, phospholipids and TAGs[65]. Moreover, there is direct and indirect contact with nucleus and Golgi organelles[66].
Besides energy supply and membrane synthesis, LDs play additional roles to ensure proper cell functionality under stress. Prolonged nutrient deprivation upregulates autophagy, causing breakdown of proteins and membranous organelles, which release amino acids and lipids potentially toxic for the cell. In this sense, LDs store neutral lipids, inert within its structure[67]. Additionally, LDs serve as extra source of lipids for FAO under nutrient stress[31,68,69] and hypoxic stress[68]. LDs also ensure the maintenance of redox homeostasis, proper mitochondrial function and membrane and organelle homeostasis[64,70]. In addition, they protect against ER stress; that is, against imbalances in ER protein folding capacity, calcium uptake and lipid composition[41,71]. Finally, LDs produce lipid intermediates that include pro- and anti-inflammatory signalling molecules[72].
LDs IN CANCER STEMNESS AND CHEMORESISTANCE
Considering LDs regulate different cellular processes, it is not surprising that they have been strongly associated to cancer progression and aggressiveness in recent years[69,73-75]. In fact, LDs facilitate not only tumour growth, but also metastasis, chemoresistance and disease relapse in multiple types of cancers[68,74,76], all processes intimately related to CSCs.
Indeed, a direct relation between LD content and stemness has been demonstrated in different types of cancers such as pancreatic, colorectal, ovarian and breast cancer[25,77-79]: On the one hand, the isolation of cells with high LD content led to an enrichment of CSCs; on the other hand, isolated CD133+CSCs show higher LD content than differentiated CD133-cancer cells. Interestingly, tumour-initiating pancreatic cells resistant to KRAS ablation showed an LD accumulation coupled with macrolipophagy, corresponding to the fusion of LD with autophagosomes. Correlated with a high catabolism rate of endogenous lipids and FAs, Vialeet al[80] determined that these KRAS ablation-resistant cells used autophagy/macrolipophagy to maintain their energy balance. Indeed, the inhibition of either autophagy or entry of FAs in the mitochondria (using bafilomycin or etomoxir, respectively) dramatically reduced cellular oxygen consumption rate. This metabolic stress was associated with a strong decrease of survival and sphere formation capacity[80]. Functionally, Tirinatoet al[81] demonstrated that sorted colorectal CSC with high or low LD content were able to form tumours after subcutaneous injection in immunocompromised mice, although cells with low LD content generated delayed small tumours less frequently. These results suggested that cells with high LD content increase tumorigenic potential, while cells with low LD content represented a more differentiated and less tumorigenic population. Thereby, LD content seems directly linked to tumorigenicity and is suggested as a marker of CSCs, in addition to molecular markers[81]. Moreover, LDrelated proteins from the PLINs and CIDE families can be associated to tumorigenicity in several cancer types[82]. Nevertheless, Caoet al[82] highlighted that an increased expression of PLIN2 was associated with a better survival rate in clear cell renal cell carcinoma (ccRCC), decreased with a higher tumour grade. Indeed, PLIN2 knockdown enhanced proliferation, migration and invasion of ccRCC cells. These findings underpin that more studies are needed to clearly identify the specific roles of LDassociated proteins in tumorigenesis or tumour progression, which may be cell or context-specific.
LDs seem to be necessary for CSCs functionality[40], not only to sustain energy demands and biomass production but also to regulate several important oncogenic signalling pathways such as Wnt/β-catenin and Hippo/Yes-associated protein 1 pathways[79] (Figure 2). In this sense, the PPARs superfamily directly associates signalling with LDs, since most lipid-derived second messengers produced in LDs act mainly through these nuclear receptors. Recently, Kuramotoet al[77] demonstrated that PPARα was activated in CSCs that accumulated LDs from pancreatic and colorectal cancer. At the same time, PPARα induced the expression of lipolytic factors like ATGL, leading to the release of FAs that supported stemness characteristics in a positive feedback loop. Indeed, a decreased PPARα activity, by using inhibitors or siRNAs, reduced sphere formation as well as pluripotency-related genes expression(SOX2,OCT4andNANOG) in pancreatic cellsin vitro[77]. These results suggest that pharmacological agents modulating PPARs activity could represent interesting compounds in order to target CSCs.
Several studies have demonstrated the importance of LDs and the associated lipase HSL in invasion and metastasis regulation, with special relevance in pancreatic cancer[83]. For instance, oncogenic KRAS down-regulates HSL to control lipid storage and utilisation, leading to LD accumulation and tumour invasion[84,85]. Disruption of the KRAS-HSL axis or overexpression of HSL reduces lipid storage and suppresses invasive migrationin vitroand metastasisin vivo[83,84]. Interestingly, Mitraet al[86] demonstrated by Raman spectrometry that circulating tumour cells isolated from the peripheral blood of patients with metastatic prostate cancer, accumulated LDs[86], further strengthening the relation between metastasis and LD accumulation.
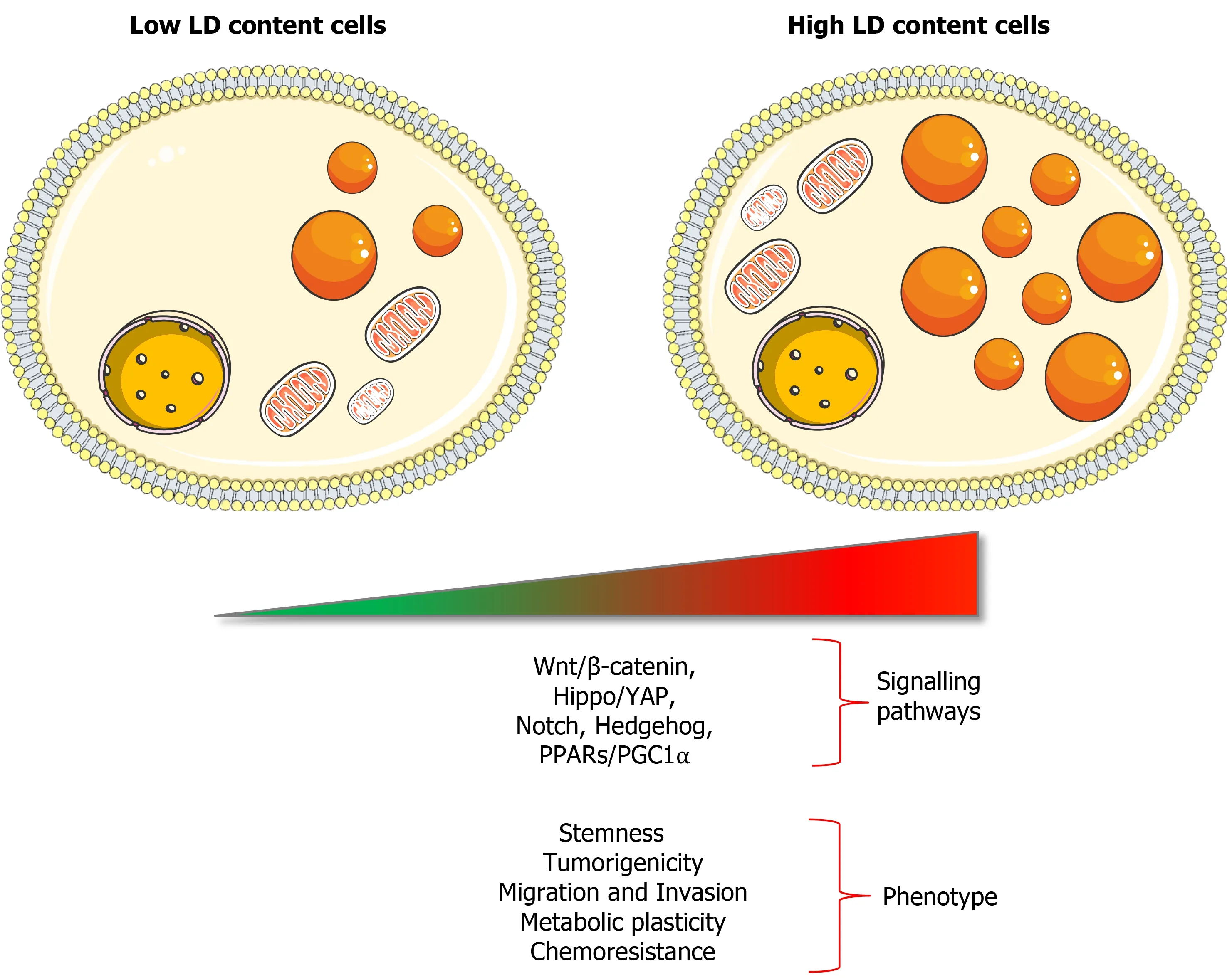
Figure 2 Features of cells with high content of lipid droplets.
Increasing evidence links lipid metabolism with chemoresistance in different cancer types[74]. For instance, FAO-derived adenosine triphosphate has been shown to drive chemoresistance in breast cancer and leukemic stem cells[87,88]. In addition, Incioet al[89] showed that 5-Fluorouracil (5-FU) uptake and efficacy in pancreatic cancer cells decreased significantly in an obese context, indicating that large obesity-caused accumulation of LDs resulting from obesity can reduce drug delivery and chemotherapy efficiency.
The contribution of LDs to chemoresistance is twofold: On the one hand, intrinsic presence of LDs has been widely reported to be a characteristic of chemoresistant cancer cell lines[68,69,74,76]. For instance, prostate cancer cells survive androgen deprivation therapy by metabolising lipids present in LDs[90]. On the other hand, chemotherapy treatments may inducede novoLD biogenesis. For example, doxorubicin and 5-FU induced TAG biosynthesis, accumulated in LDs in human colon carcinoma cells[74,91]. Moreover, direct or indirect pharmacological inhibition of FAO or OXPHOS is sufficient to drive LD formation in cancer cells[74]. Indeed, treatment with the c-MYC/Max inhibitor 10058-F4 induced LD accumulation resulting from mitochondrial dysfunction[92]. Interestingly, a combination of both LD presence and accumulation has been described in colorectal cancer cells. For instance, high LD content identified cancer cell lines with increased chemoresistance to 5-FU and oxaliplatin. These cells further accumulated LDs in response to chemotherapy in a process facilitated by lysophosphatidyl-choline acyltransferase 2 (LPCAT2), an LDassociated enzyme essential for phosphatidylcholine synthesis[93]. An elevated expression of LPCAT2 prevented chemotherapy-induced ER stress, further highlighting the protective role of LDs against cellular stresses[74,93]. Importantly, it has been recently reported that LDs can also act as a sink to sequester hydrophobic compounds impairing drug-induced apoptosis, resulting in chemoresistance of cancer cells[68,69].
CONCLUSION
Even if our knowledge about the mechanisms by which LDs support cancer stemness is still very limited, it seems clear now that high levels of LDs are strongly associated with cancer aggressiveness and chemotherapy resistance in different tumour types. Considering this, measurement of LD accumulation could be potentially used as a prognostic biomarker, also with predictive value in terms of treatment response to conventional therapies. A deeper understanding of the molecular mechanisms dictating their implication in essential processes of the CSC biology, such as tumorigenicity, metastatic spread and chemoresistance, should pave the way to discover novel LD-related targets and therapeutic approaches for more effective cancer treatment.
ACKNOWLEDGEMENTS
We want to thank Laura Sancho for proofreading the manuscript.
杂志排行

World Journal of Stem Cells的其它文章
- Effects of living and metabolically inactive mesenchymal stromal cells and their derivatives on monocytes and macrophages
- Stem cells' centrosomes: How can organelles identified 130 years ago contribute to the future of regenerative medicine?
- Effects of storage media, supplements and cryopreservation methods on quality of stem cells
- Recent advances in stem cell therapy for neurodegenerative disease: Three dimensional tracing and its emerging use
- Stem cell therapies in cardiac diseases: Current status and future possibilities
- Current evidence on potential of adipose derived stem cells to enhance bone regeneration and future projection