K助剂不同形态在MoP(101)表面吸附的理论研究
2021-09-30田欣欣
田欣欣,李 盼
(山西大学 分子科学研究所,山西 太原 030006)
合成气(CO和H2的混合气)来源丰富,可由化石燃料(煤和天然气)、生物质或有机废弃物等多种含碳物质制备[1-2].目前,按照原料与工艺的不同,制备合成气的方法可分为煤气化、天然气气化、天然气部分氧化、天然气蒸汽重整和生物质气化等[3].合成气转化得到的产物主要包括烃类、甲醇和低碳醇等多种化合物.其中,低碳醇是生产多种化学品的原料和中间体,广泛应用于制备药物、塑料和表面活性剂等行业[4-7].此外,它也是一种理想的燃油添加剂和新型燃料替代品.
合成气直接转化生成低碳醇的整个反应过程非常复杂,除了生成低碳醇这一主要产物,催化剂表面也会同时发生其他副反应,导致诸如烃类、水以及酸、酯、醚等多种含氧化合物的生成.目前,普遍认为合成气转化为低碳醇的反应机理是双功能活性中心协同催化,即一种活性位点催化C-C耦合反应,实现碳链的增长,另一种活性位点催化CO分子的吸附与加氢反应[8-11].因此,单一金属作催化剂通常没有较好的催化活性.但可以通过加入碱金属助剂和载体进一步调控催化剂的电子与几何性质,提高催化活性.
目前合成气直接转化制备低碳醇的催化剂体系主要分为2种,即贵金属Rh基催化剂和非贵金属催化剂.其中,非贵金属催化剂又包括改性甲醇合成催化剂、改性费托(F-T)合成催化剂和Mo基催化剂这3种.其中,MoP是一种类贵金属催化剂,其具有成本低、抗硫毒化能力强和加氢活性良好等优点.与其他Mo基催化剂相比,掺杂碱金属K助剂的MoP催化剂对烃类的选择性最低,对C2+含氧化合物的选择性最高[12-13].Zaman和Smith[12-14]对K/MoP催化体系催化合成气转化进行了系统地研究,他们发现在此体系中添加K助剂后,产物的选择性发生了改变,碳氢物种的形成受到抑制,但C2+含氧化合物的选择性增大.为了进一步提高低碳醇的选择性,Xu等[15]制备了一系列KxCo0.75MoP (0≤x≤1.5)催化剂.结果表明,K/Co比例为K0Co0.75和K1Co0.75时,CH4的选择性分别为30.5%和13.3%,对C2+含氧化合物的选择性分别为20.4%和43.2%,即加入适量的K助剂抑制了CH4的生成,并提高了C2+含氧化合物的选择性,这与Zaman等发现的趋势是一致的.XPS表征发现对于 KxCo0.75MoP (x=0、1、1.5),Mo4+的含量分别是0%、100%和100%,因此,他们认为K助剂促进了不同Mo物种的电子重排,高含量Mo4+是提高低碳醇选择性的原因.最近,Jaramillo等[16]系统地研究了K助剂对MoP催化合成气转化的影响.他们也发现加入适量K,低碳醇的选择性得到提高,而CH4的生成受到抑制,当K含量过高时,低碳醇的选择性反而降低.
尽管已有诸多报道表明K助剂在合成气转化生成低碳醇反应中具有良好的促进效果,但是目前关于其作用机理的理论研究尚不全面.Zaman等[17]在探究K助剂作用机理的理论研究中使用的模型为Mo6P3团簇.但该模型尺寸过小,不能代表实际反应过程中不同尺寸催化剂表面的电子性质.同时,由于对MoP催化剂表面稳定性顺序以及表面性质缺乏系统地认识,有关表面机理的理论研究鲜有报道.2018年,Tian等[18]系统计算了MoP表面不同终结的表面能,发现(101)是最稳定表面,且(101)表面在MoP的Wulff结构中暴露比例最大.这一结果为机理研究中表面模型的选择提供了参考.另外对(101)表面CO和H2共吸附的研究也表明,该表面具有复杂的CO/H2共吸附比例[19],有利于合成气转化反应的发生.
基于以上研究背景,本文采用密度泛函理论研究了K助剂在MoP(101)表面上的吸附情况,分析和对比了K原子态和K2O分子态吸附对该表面几何结构和电子结构的影响,揭示了K助剂与MoP(101)表面的作用机理,为后续的表面机理研究奠定了理论基础.
1 计算方法与模型
本文所有密度泛函理论(DFT)计算均采用VASP软件包完成[20-21].使用投影缀加波(PAW)赝势描述核电子与原子核间的相互作用[22].方法采用广义梯度近似(GGA) PBE泛函[23],同时采用DFT-D3色散校正考虑弱相互作用的影响[24].截断能设置为 450 eV.结构优化过程中,力和能量的收敛标准分别设为 0.2 eV/nm 和10-4eV.本文中所有的能量均进行了零点能校正.
MoP晶体结构模型选择最稳定的WC-型[25-26],空间群为P-6m2[27].优化后的晶格参数为a=b=0.323 9 nm, c=0.319 3 nm,与实验值相吻合(a=b=0.322 3 nm, c=0.319 1 nm)[28].选用MoP(101)表面4层厚p(2×3)的超胞为模型进行相关计算,允许模型上面2层原子和吸附物种弛豫,并固定下面2层原子来模拟催化剂体相.为了避免周期性模型层间相互作用的影响,真空层厚度设置为 1.5 nm.计算采用3×3×1的布里渊区k点.
吸附物种在催化剂表面的吸附能Eads定义为:
Eads=Etotal-Eslab-Eadsorbate
其中,Etotal代表物种吸附在MoP(101)表面时的总电子能量,Eslab代表洁净MoP(101)表面的总电子能量,Eadsorbate代表吸附物种在气相中的电子能量.由此可见,吸附能数值越负,吸附过程放热越多,则吸附构型越稳定.
2 结果与讨论
2.1 K原子吸附
2.1.1 吸附结构与吸附能
研究了K原子在MoP(101)表面不同位点的吸附情况.通过几何优化,发现在MoP(101)表面K原子共有2种不同的稳定吸附位点,分别为洞位H和桥位B.而吸附于其它位点上的K原子在优化过程中均移动到更加稳定的H位.表1列出了K原子吸附在MoP(101)表面的吸附能、键长和bader电荷.
由表1可知,K原子在MoP(101)表面的吸附能为负值,表明K原子在MoP(101)表面能形成稳定的化学吸附.比较不同位点的吸附能可知,K原子在H位的吸附能最负,吸附最稳定.因此,在后续电子结构的计算中,只考虑K原子在MoP(101)表面H位上的吸附.此外,bader电荷分析表明K原子吸附在MoP(101)表面后,向表面转移了0.79个电子.
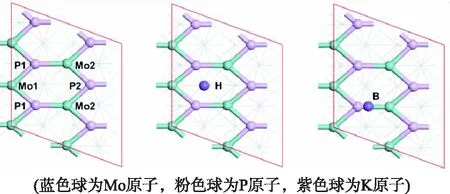
图1 MoP(101)表面结构及K原子的稳定吸附位点图

表1 MoP(101)表面K原子吸附的吸附能、键长和bader电荷
2.1.2 电子结构
为了进一步理解K原子吸附对MoP(101)表面性质的影响,计算了MoP(101)和K/MoP(101)表面功函数.
如表2所示,与洁净MoP(101)表面相比,加入K原子后MoP(101)表面功函数明显降低,这表明K原子主要通过电子效应影响MoP(101)面的表面性质.加入K原子后,MoP(101)表面变得更加活泼,易于发生化学反应.
为了研究K原子吸附前后电子分布的变化,也计算了洁净MoP(101)表面的态密度以及K/MoP(101)表面吸附体系的态密度,以洁净MoP(101)表面的费米能级作为能量零点.
图2为洁净MoP(101)表面的总态密度、Mo原子和P原子的分波态密度.在态密度图中,-13.5~-10 eV 能量区间的能带,主要是P原子3s轨道的贡献.价带在-7~0 eV 能量区间,主要是Mo的4d轨道和P的3p轨道的贡献,且Mo的4d轨道对费米能级附近的电子结构影响较大.导带在0~4 eV 能量区间,主要是Mo的4d轨道的贡献.
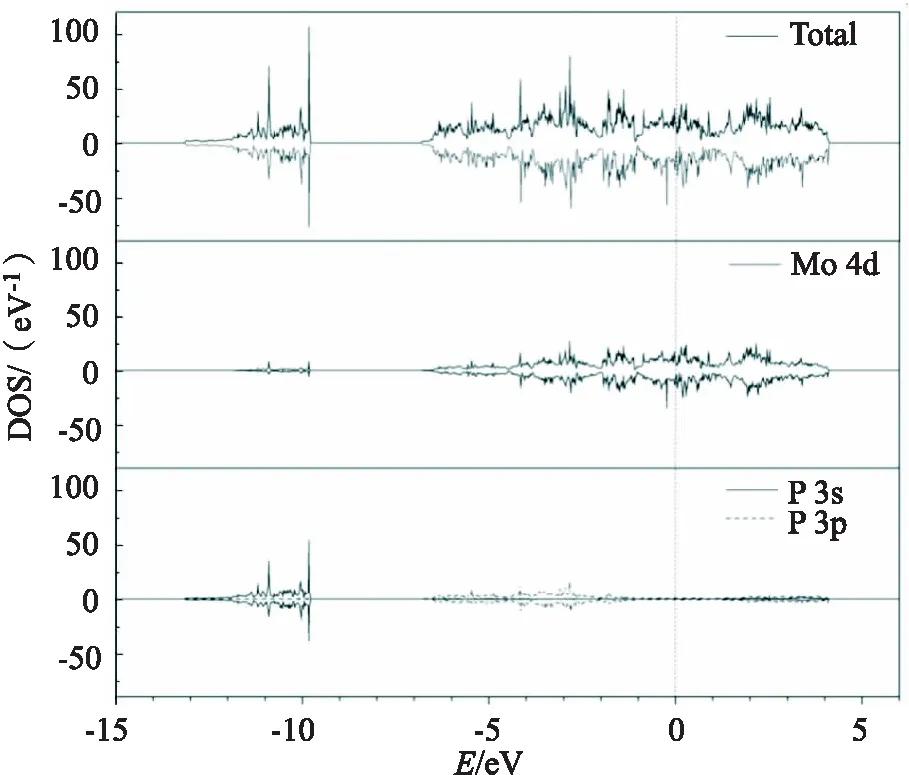
图2 MoP(101)表面态密度
K原子吸附于MoP(101)表面的总态密度、Mo原子、P原子和K原子的分波态密度如图3所示.吸附K原子后MoP(101)表面的总态密度整体形状未发生明显变化,价带在-6~0 eV 能量区间,主要是Mo的4d轨道和P的3p轨道的贡献,且Mo的4d轨道对费米能级附近的电子结构影响较大;导带在0~5.5 eV 能量区间,主要是Mo的4d轨道的贡献,表明吸附K原子后价带和导带均向右(高能量区域)移动.此外,在-11~-9 eV 范围内,K的3p轨道与P的3s轨道以及Mo的4d轨道发生重叠,存在较强的相互作用;在-6~-1 eV 范围内,K的3p/4s轨道与P的3p轨道以及Mo的4d轨道发生重叠,存在较强的相互作用.吸附K原子的态密度对导带的影响较大,其部分3p和4s电子处于费米能级上方0~5.5 eV范 围内,为未占据态.这表明电荷从K原子转移到MoP(101)表面,这与bader电荷分析得到的结果一致.
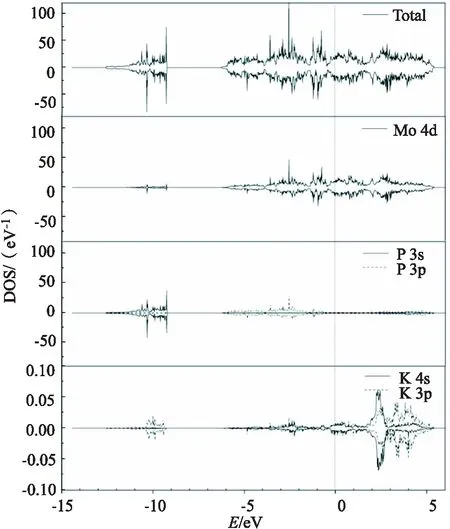
图3 K/MoP(101)表面态密度
2.2 K2O吸附
2.2.1 吸附结构与吸附能
研究了K2O在MoP(101)表面不同位点的吸附情况.通过几何优化,发现在MoP(101)表面K2O有多种不同的稳定吸附位点,其中最稳定的3种吸附构型如图4所示,分别为顶位T1和T2以及桥位B.表2列出了K2O在MoP(101)表面的吸附能、键长、键角和bader电荷.
由表2可知,K原子在MoP(101)表面的吸附能为负值,表明K2O在MoP(101)表面能形成稳定的化学吸附.比较不同位点的吸附能可知,K2O在T1位的吸附能最负,吸附最稳定.因此,在后续电子结构的计算中,只考虑K2O在MoP(101)表面T1位上的吸附.此外,bader电荷分析表明K2O吸附在MoP(101)表面后,向表面转移了0.49个电子.
2.2.2 电子结构
同样地,为了进一步理解K2O吸附对MoP(101)表面性质的影响,计算了K2O/MoP(101)表面功函数,发现加入K2O后,表面功函数降为 1.64 eV(费米能级为 2.64 eV,静电势能为 4.28 eV),甚至低于K/MoP(101)表面.
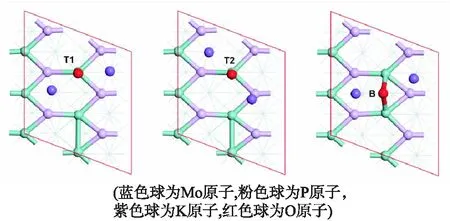
图4 K2O在MoP(101)表面的稳定吸附位点图
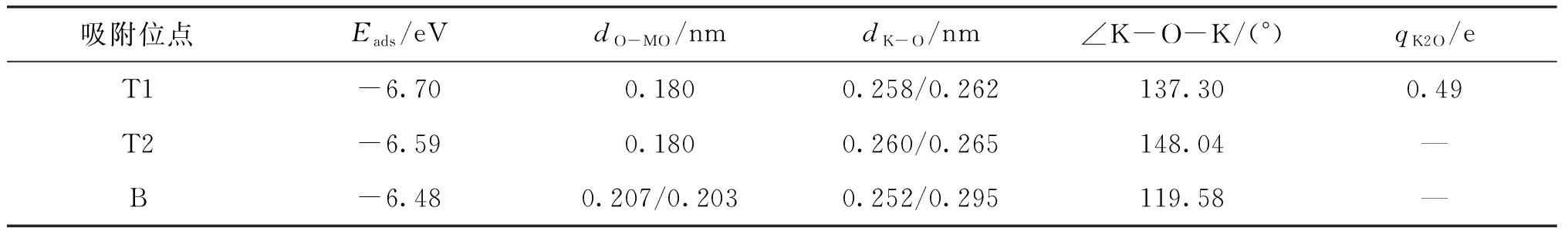
表3 K2O吸附于MoP(101)表面的吸附能、键长、键角和bader电荷
为了研究K2O吸附前后电子分布的变化,也计算了K2O/MoP(101)表面吸附体系的态密度(图5).以洁净MoP(101)表面的费米能级为能量零点,K2O吸附于MoP(101)表面的总态密度、Mo原子、P原子、K原子和O原子的分波态密度如图4所示.可知,吸附K2O后MoP(101)表面的总态密度整体形状未发生明显变化.吸附后的价带在-6~0 eV 能量区间,主要是Mo的4d轨道和P的3p轨道的贡献,且Mo的4d轨道对费米能级附近的电子结构影响较大;导带在0~5.5 eV 能量区间,主要是Mo的4d轨道贡献,表明吸附后价带和导带均向右(高能量区域)移动.在 -13~-9 eV 范围内,K的3p轨道与P的3s轨道以及Mo的4d轨道发生重叠,存在较强的相互作用;在-6~0 eV 范围内,K的3p/4s轨道与P的3s轨道以及Mo的4d轨道发生重叠,存在较强的相互作用.此外,K原子的部分3p和4s电子态处于费米能级上方0~5.5 eV 范围内,变为未占据态.这表明电荷从K原子转移到MoP(101)表面,与bader电荷分析得到的结果一致.与K/MoP(101)表面K原子的态密度相比,K2O中K原子的态密度对价带的贡献增大,这可能是由于分子态K2O中有2个K原子,但相比于Mo的4d轨道,K的贡献仍然小.
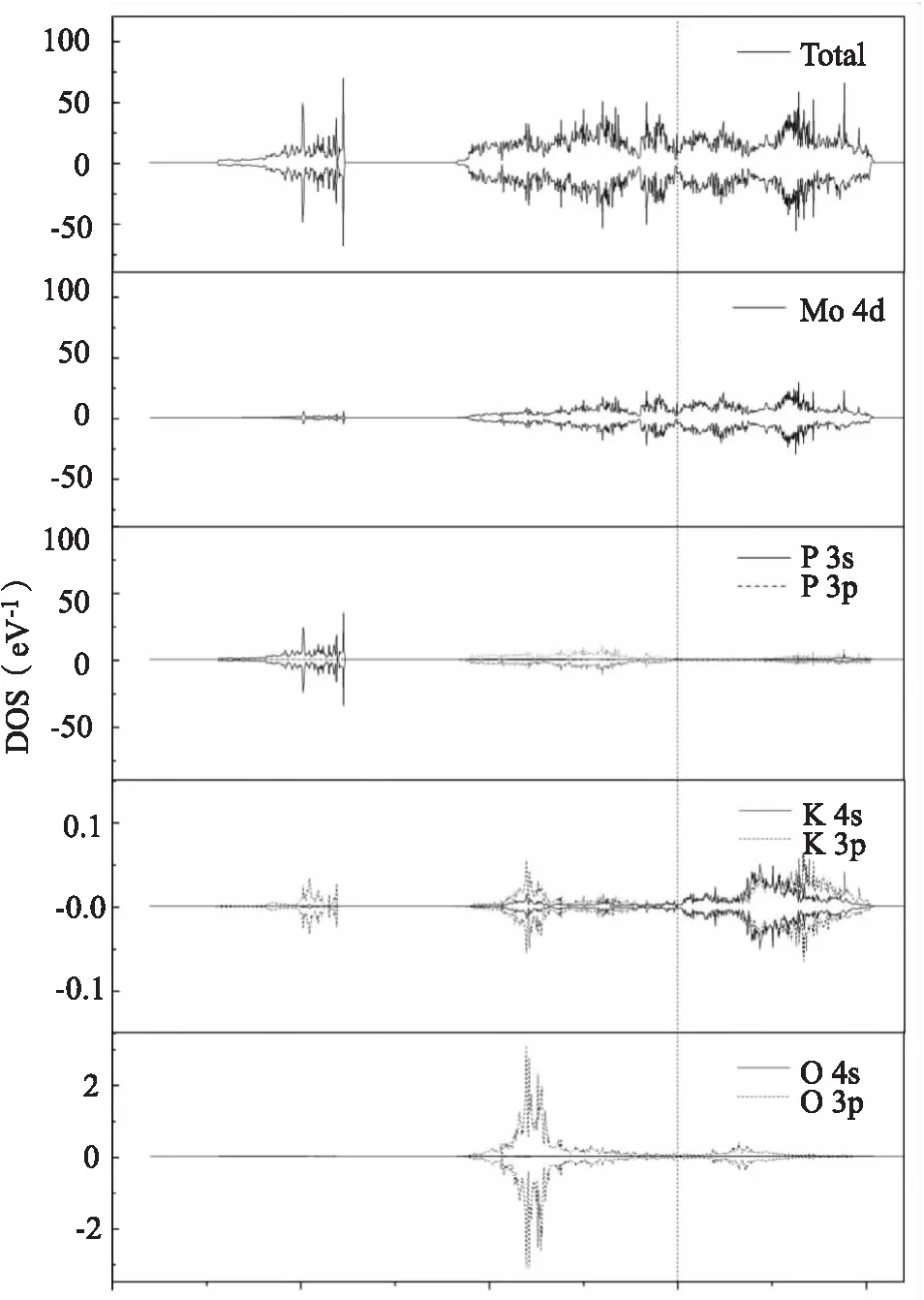
图5 K2O/MoP(101)表面态密度
3 结语
采用密度泛函理论,研究了K助剂以不同形态吸附于MoP(101)表面的稳定构型以及电子结构.研究发现无论是原子态吸附的K原子还是分子态吸附的K2O均会向MoP(101)表面转移电子,使其功函数明显降低,态密度分析表明,K原子和K2O均与MoP(101)表面均存在较强的相互作用.整体来看,MoP(101)表面吸附K和K2O后,价带和导带均向高能量区域移动,但K和K2O引起的变化非常接近.因此,可猜测,K助剂的不同形态对表面电子性质的影响是相近的,在后续的表面机理研究中,为了简化模型和节省计算量,可以采用K原子吸附的模型来进行相关定性研究.
