MEG3-miR-455-PI3K/Akt信号通路对大脑中动脉闭塞小鼠模型缺氧损伤神经干细胞的影响
2021-09-24白瑞瑞于奇晋李定安
王 衡,陈 蓉,白瑞瑞,于奇晋,姜 进,李定安
脑梗死是危害人类健康的重要原因之一,而神经干细胞(neural stem cells,NSCs)的增殖及分化在脑梗死等缺血性脑血管疾病中发挥重要作用,缺氧时间的延长可促进NSCs凋亡[1]。目前关于NSCs在缺氧中的作用机制尚未阐明。长链非编码RNA(long non-coding RNA,LncRNA)在神经系发育过程中发挥重要调控作用,研究表明LncRNA可调控神经细胞分化过程[2-4]。LncRNA母系表达基因3(LncRNA MEG3)在神经细胞缺血缺氧模型中表达升高,母系表达基因3(MEG3)可促进缺血缺氧造成的小鼠神经母细胞瘤细胞凋亡[5]。但MEG3对小鼠脑梗死缺氧神经干细胞损伤的影响尚未可知。生物信息学分析显示微小RNA-455(microRNA-455,miR-455)可能是MEG3的靶基因,研究表明,miR-455在氧糖剥夺神经细胞和大脑中动脉闭塞(middle cerebral artery occlusion,MCAO)小鼠脑组织中呈低表达,并可抑制缺血性脑卒中的神经细胞凋亡[6]。相关报道指出,磷脂酰肌醇激酶(phosphoinositide 3-kinase,PI3K)/蛋白激酶 B(protein kinase B,Akt)信号通路可能与脑梗死大鼠脑损伤、血管新生和氧化应激有关[7]。本研究通过构建MCAO小鼠模型,观察MEG3、miR-455在MCAO模型中的表达水平,原代分离与培养NSCs并进行缺氧处理构建NSCs缺氧损伤模型,检测细胞中MEG3与miR-455的表达水平,探究MEG3表达变化对NSCs增殖、分化、凋亡及氧化应激的影响,初步分析其对miR-455及PI3K/Akt信号通路的调控作用,以揭示NSCs缺氧损伤机制。
1 材料与方法
1.1 实验动物 无特定病原体(SPF)级健康雄性C57BL/6小鼠36只,购自上海斯莱克实验动物有限公司,动物许可证号为SCXK(沪):2016-0003。
1.2 实验材料 杜氏改良培养基(DMEM)、胎牛血清购自美国Gibco公司;胰蛋白酶购自美国Thermo Fisher公司;Lipofectamine2000与Trizol试剂购自美国Invitrogen公司;反转录试剂盒、SYBR Green试剂盒购自北京天根生化科技有限公司;MEG3小干扰RNA(si-MEG3)、乱序无意义阴性对照(si-con)、miR-455模拟物(mimics)、阴性对照(miR-con)、miR-455特异性寡核苷酸抑制剂(anti-miR-455)及其阴性对照(anti-miR-con)购自广州锐博生物科技有限公司;PI3K/Akt信号通路抑制剂LY294002购自美国Sigma公司;甲基噻唑基四唑(MTT)购自北京百奥莱博科技有限公司;细胞凋亡检测试剂盒购自北京索莱宝科技有限公司;超氧化物歧化酶(SOD)、谷胱甘肽过氧化物酶(GSH-Px)、丙二醛(MDA)检测试剂盒购自南京建成生物工程研究所;兔抗鼠CyclinD1、p21抗体购自上海善然生物科技有限公司;兔抗鼠B淋巴细胞瘤-2(Bcl-2)、B淋巴细胞瘤-2相关蛋白(Bax)抗体购自美国Abcam公司;兔抗鼠神经胶质纤维酸性蛋白(GFAP)、Ⅲ型β微管蛋白(β-tubulin-Ⅲ)抗体购自北京百奥莱博科技有限公司;兔抗鼠磷酸化磷脂酰肌醇激酶(p-PI3K)、磷酸化蛋白激酶 B(p-AKT)购自美国GeneTex公司;兔抗鼠PI3K、Akt抗体购自美国Epitmics公司;辣根过氧化物酶(HRP)标记的山羊抗兔二抗购自武汉艾美捷科技有限公司;RIPA(radio immunoprecipitation assay)裂解液、二喹啉甲酸(BCA)蛋白定量检测试剂盒、增强型化学发光试剂(ECL)、二烷基硫酸钠(SDS)购自北京全式金生物技术有限公司。
1.3 实验方法
1.3.1 MCAO模型建立 将18只大鼠随机分为假手术组与MCAO组,每组9只。MCAO模型:使用65 mg/kg的氯胺酮与6 mg/kg的甲苯噻嗪经腹腔注射入小鼠体内行急性麻醉处理,参照相关文献制备MCAO模型,以缺血期血流降至基线水平25%以下且灌注期血流恢复至80%以上视为建模成功[8]。假手术组:除不用尼龙线栓外,其余步骤同MCAO组。切除各组小鼠全脑组织,置于-80 ℃超低温冰箱保存。
1.3.2 NSCs分离及培养 取18只小鼠(月龄6周),采用脱颈法处死小鼠,75%乙醇消毒,取小鼠大脑,剔除脑膜等结缔组织,盐溶液清洗,将脑组织剪成1 mm3的碎块,加入0.25%胰蛋白酶消化15 min,采用200目筛网过滤,4 ℃条件下经1 000 r/min转速离心10 min,弃上清,收集细胞,参照相关文献进行培养及传代,收取第4代细胞进行后续实验[9]。
1.3.3 建立缺氧NSCs损伤模型及实验分组 收集NSCs接种于无糖DMEM培养基(5×105个/mL),置于缺氧盒内,通入5% CO2与95% N2混合气体1 h后,关闭气体,取出细胞[10]。将正常培养的NSCs作为对照组,缺氧处理的NSCs作为模型组。取对数生长期NSCs,分别将si-MEG3、si-con、si-MEG3与 anti-miR-455共转染至NSCs,转染步骤参照Lipofectamine 2000说明书进行操作,转染48 h后进行缺氧处理,分别作为Hypoxia+si-MEG3组、Hypoxia+si-con组、Hypoxia+si-MEG3+anti-miR-455组。为探究MEG3对PI3K/Akt信号通路的调控作用,分别将si-MEG3转染至NSCs后使用浓度为50 μmol/L的LY294002处理24 h[11],随后缺氧处理,作为Hypoxia+si-MEG3+LY294002组。为探究MEG3是否通过靶向miR-455调控PI3K/Akt信号通路,分别将si-con、si-MEG3、si-MEG3与anti-miR-con、si-MEG3与anti-miR-455共转染至NSCs,分别记作si-con组、si-MEG3组、si-MEG3+anti-miR-con组及si-MEG3+anti-miR-455组,转染方法同上。
1.3.4 实时荧光定量聚合酶链反应(quantitative real-time PCR,qRT-PCR)检测细胞中MEG3、miR-455表达水平 采用Trizol法提取假手术组、MCAO组、对照组、模型组及转染组NSCs中的总RNA,应用Nanodrop 2000 c超微量分光光度计检测RNA浓度。参照反转录试剂盒将RNA反转录合成cDNA。qRT-PCR扩增反应体系:SYBR Green Master Mix 10 μL,正向、反向引物0.8 μL,cDNA 2 μL,ddH2O补足体系至20 μL;反应条件:95 ℃预变性5 min循环1次,95 ℃变性15 s,60 ℃退火60 s,72 ℃延伸30 s,共循环40次。MEG3以β-actin为内参,miR-455以U6为内参,采用2-ΔΔCt法计算MEG3、miR-455相对表达量。
1.3.5 MTT检测细胞增殖 收集各组NSCs接种于96孔板,按照1.3.3中程序分组处理,每组设置3个复孔,加入质量浓度为5 mg/mL的MTT溶液每孔20 μL,放入37 ℃恒温培养箱培养4 h,4 ℃条件下经1 300 r/min离心5 min,弃上清,加入二甲基亚砜(DMSO)每孔150 μL,室温避光振荡、孵育5 min,应用酶标仪检测各孔吸光度值。
1.3.6 流式细胞术检测细胞凋亡率 收集各组NSCs,0.25%胰蛋白酶消化后离心1 000 r/min,离心6 min,弃上清,加入磷酸缓冲盐溶液(PBS)洗涤,加入500 μL结合缓冲液,充分混匀,加入5 μL 膜联蛋白V(Annexin V)-异硫氰酸荧光素(FITC),充分混匀,加入5 μL碘化丙啶(PI),充分混匀,于1 h内置于FACS Calibur流式细胞仪及应用Cellauest软件检测各组细胞凋亡率。
1.3.7 SOD、GSH-Px活性与MDA含量检测 收集各组NSCs培养上清液,按照检测试剂盒分别检测SOD、GSH-Px活性与MDA含量,严格按照试剂盒说明书进行操作。
1.3.8 双荧光素酶报告基因检测MEG3与miR-455的靶向关系 经LncBase网站预测LncRNA MEG3和miR-455存在靶向结合位点,分别构建野生型载体WT-MEG3与突变型载体MUT-MEG3,取生长状态良好的NSCs,将WT-MEG3、MUT-MEG3分别与miR-con、miR-455 mimics共转染至NSCs,将其作为miR-con组及miR-455组,转染48 h后收集细胞,检测各组相对荧光素酶活性。
1.3.9 蛋白免疫印迹法(Western Blot)检测CyclinD1、Bcl-2、p21、Bax、GFAP、β-tubulin-Ⅲ与PI3K/Akt信号通路相关蛋白表达 收集各组对数期NSCs,加入RIPA裂解液,冰浴30 min,4 ℃条件下经1 000 r/min转速离心10 min,吸取上清液。采用BCA法测定蛋白浓度,取30 μg蛋白上样量加入SDS上样缓冲液,沸水中煮10 min。取蛋白样品进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE),将电泳产物转移至聚偏二氟乙烯(PVDF)膜,封闭,加入一抗稀释液,10×封闭-洗涤缓冲液(TBST)洗涤,4 ℃孵育24 h,TBST洗涤,孵育二抗稀释液,室温孵育1 h,滴加ECL,暗室内曝光显影,应用凝胶成像分析系统及Image J软件分析各条带灰度值。
2 结 果
2.1 MEG3和miR-455在MACO小鼠脑组织和缺氧NSCs损伤模型中的表达情况 qRT-PCR检测结果显示,与假手术组比较,MACO组小鼠脑组织中MEG3表达水平明显升高(P<0.05),miR-455表达水平明显降低(P<0.05)。在缺氧NSCs损伤模型中,与对照组比较,模型组MEG3表达水平明显升高(P<0.05),miR-455表达水平明显降低(P<0.05)。详见图1、图2。

与假手术组比较,*P<0.05。

与对照组比较,*P<0.05。
2.2 抑制MEG3表达对缺氧NSCs损伤模型细胞增殖、凋亡及凋亡相关蛋白表达的影响 与对照组比较,模型组MEG3表达水平明显升高(P<0.05),细胞增殖活力明显降低(P<0.05),细胞凋亡率明显升高(P<0.05),CyclinD1、Bcl-2蛋白相对表达量明显降低(P<0.05),p21、Bax蛋白相对表达量明显升高(P<0.05);与Hypoxia+si-con组比较,Hypoxia+si-MEG3组MEG3表达水平明显降低(P<0.05),48 h、72 h细胞增殖活力明显升高(P<0.05),细胞凋亡率明显降低(P<0.05),CyclinD1、Bcl-2蛋白相对表达量明显升高(P<0.05),p21、Bax蛋白相对表达量明显降低(P<0.05)。详见图3、图4及表1。

图3 抑制MEG3表达对缺氧NSCs损伤模型细胞凋亡的影响

与对照组比较,*P<0.05;与Hypoxia+si-con组比较,# P<0.05。
表1 各组MEG3表达、细胞增殖活力、细胞凋亡率比较(±s)
2.3 抑制MEG3表达对缺氧NSCs损伤模型细胞分化和氧化应激水平的影响 与对照组比较,模型组细胞中GFAP、β-tubulin-Ⅲ蛋白相对表达量明显降低(P<0.05),SOD、GSH-Px活性明显降低(P<0.05),MDA活性明显升高(P<0.05);与Hypoxia+si-con组比较,Hypoxia+si-MEG3组细胞中GFAP、β-tubulin-Ⅲ蛋白相对表达量明显升高(P<0.05),SOD、GSH-Px活性明显升高(P<0.05),MDA含量明显降低(P<0.05)。详见图5及表2、表3。
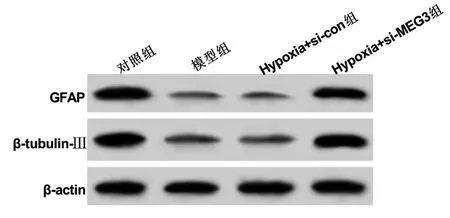
图5 抑制MEG3表达对缺氧NSCs损伤模型GFAP和 β-tubulin-Ⅲ蛋白表达影响的条带图
表2 抑制MEG3表达对缺氧NSCs损伤模型GFAP和 β-tubulin-Ⅲ蛋白表达的影响(±s)
表3 抑制MEG3表达对缺氧NSCs损伤模型氧化应激指标水平的影响(±s)
2.4 MEG3与miR-455的靶向关系 经LncBase网站预测显示MEG3和miR-455存在结合位点(见图6)。双荧光素酶报告实验检测结果显示,转染野生型报告基因载体WT-MEG3的细胞中,与miR-con组(1.00±0.10)比较,miR-455组(0.38±0.07)荧光素酶活性明显降低(t=15.238,P<0.05);转染突变型报告基因载体MUT-MEG3的细胞中,与miR-con组(1.02±0.12)比较,miR-455组(0.99±0.13)荧光素酶活性无明显变化(t=0.509,P=0.618)。qRT-PCR检测结果显示,与si-con组(1.00±0.15)比较,si-MEG3组(3.17±0.21)细胞中miR-455的表达水平明显升高(t=25.226,P<0.05)。

图6 MEG3与miR-455的结合位点
2.5 MEG3通过靶向miR-455调控PI3K/Akt信号通路 Western Blot检测结果显示,与si-con组比较,si-MEG3组细胞中p-PI3K、p-Akt蛋白相对表达量明显升高(P<0.05);与si-MEG3+anti-miR-con组比较,si-MEG3+anti-miR-455组细胞中p-PI3K、p-Akt蛋白相对表达量明显降低(P<0.05),不同组间PI3K、Akt蛋白相对表达量比较差异均无统计学意义(P>0.05)。详见图7。

与si-con组比较,*P<0.05;与si-MEG3+anti-miR-con组比较,#P<0.05。
2.6 抑制miR-455或PI3K/Akt信号通路能逆转抑制MEG3对缺氧NSCs损伤模型的影响 与Hypoxia+si-MEG3组比较,Hypoxia+si-MEG3+anti-miR-455组、Hypoxia+si-MEG3+LY294002组48 h、72 h细胞增殖活力均明显降低(P<0.05),细胞凋亡率明显升高(P<0.05),CyclinD1、Bcl-2蛋白相对表达量明显降低(P<0.05),p21、Bax蛋白相对表达量明显升高(P<0.05),GFAP、β-tubulin-Ⅲ蛋白相对表达量及SOD、GSH-Px活性明显降低(P<0.05),MDA含量明显升高(P<0.05)。详见图8~图10及表4、表5。
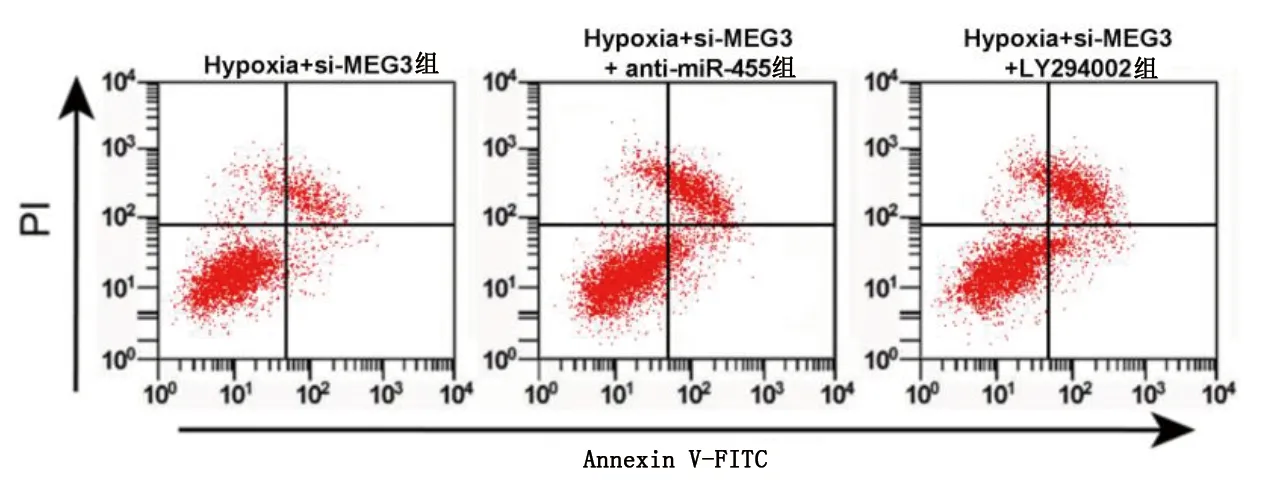
图8 抑制miR-455或PI3K/Akt信号通路能逆转抑制MEG3对缺氧NSCs损伤模型细胞凋亡的影响

与Hypoxia+si-MEG3组比较,*P<0.05。
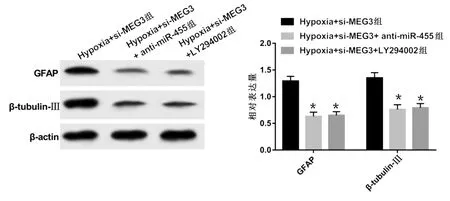
与Hypoxia+si-MEG3组比较,*P<0.05。
表4 抑制miR-455或PI3K/Akt信号通路能逆转抑制MEG3对缺氧NSCs损伤模型细胞增殖、凋亡的影响(±s)
表5 抑制miR-455或PI3K/Akt信号通路能逆转抑制MEG3对缺氧NSCs损伤模型氧化应激水平的影响(±s)
3 讨 论
研究表明MEG3与糖尿病相关的微血管功能障碍有关[12],而MEG3还通过靶向miR-21/PDCD4信号通路促进缺血性神经元死亡[13],此外,沉默MEG3表达可促进神经生长并减轻大鼠脑缺血再灌注损伤[14]。本研究结果显示,MCAO组MEG3表达水平明显高于假手术组,同时模型组细胞中MEG3表达水平明显高于对照组,提示MEG3在缺氧诱导的NSCs损伤中发挥重要调控作用。有研究表明,CyclinD1可正向调控细胞周期,促进细胞增殖,p21可负向调控细胞周期,抑制细胞增殖[15]。Bcl-2、Bax蛋白参与细胞凋亡过程,Bcl-2是抗细胞凋亡蛋白,Bax是促细胞凋亡蛋白[16]。本研究结果提示,抑制MEG3表达可能通过上调CyclinD1、Bcl-2表达及下调p21、Bax表达,从而抑制缺氧诱导的NSCs凋亡,促进细胞增殖。GFAP标记星形胶质细胞,β-tubulin-Ⅲ标记神经元,通过测定不同组GFAP与β-tubulin-Ⅲ蛋白表达可反映NSCs向星形胶质细胞和神经元分化的水平[17]。本研究结果表明,抑制MEG3表达可促进缺氧诱导的NSCs分化。氧化应激与细胞凋亡能力密切相关,自由基氧化能力增强可与体内不饱和脂肪酸反应生成MDA,从而促使细胞膜功能损伤最终引发氧化应激损伤,而SOD、GSH-Px属于抗氧化酶类,其可增强机体抗氧化能力[18]。本研究结果提示,抑制MEG3表达可有效减轻NSCs氧化应激损伤并增强其抗氧化能力。
肉桂酸通过促进miR-455-3p表达及抑制HDAC2表达从而减轻小鼠颅脑损伤[19]。有研究表明,沉默LncRNA TTTY15表达可通过上调miR-455-5p表达从而减轻缺氧诱导的心肌细胞损伤[20]。相关报道指出,miR-455-5p表达下调可激活多种炎症途径进而参与多发性硬化症疾病发生过程[21]。miR-455-3p是从miR-455前体的3′端臂加工而来,miR-455-5p从miR-455前体的5′端臂加工而来,但关于miR-455在NSCs缺氧损伤过程中的作用机制尚未可知。本研究结果显示,MCAO组、模型组miR-455表达水平明显降低,进一步研究显示MEG3可靶向结合miR-455并负向调控miR-455表达,抑制MEG3表达可明显上调miR-455表达,提示抑制MEG3表达可能通过上调miR-455从而减轻缺氧NSCs损伤。有研究表明,MEG3通过抑制PI3k/Akt信号通路从而促进蛛网膜下隙出血所致的神经细胞损伤[22]。相关报道指出PI3k/Akt信号通路激活可促进缺氧条件下NSCs增殖[23]。本研究结果显示,抑制MEG3表达可通过上调miR-455表达而激活PI3K/Akt信号通路。为验证MEG3是否通过调控miR-455与PI3K/Akt信号通路而参与NSCs增殖、分化及氧化应激过程,本研究对miR-455表达或PI3K/Akt信号通路进行抑制,结果发现细胞增殖活力降低,细胞凋亡率升高,表明抑制miR-455或PI3K/Akt信号通路能逆转抑制MEG3对缺氧损伤NSCs的影响,提示抑制MEG3表达可能通过上调miR-455表达及激活PI3K/Akt信号通路对NSCs发挥保护作用。
综上所述,MEG3表达量升高可加重NSCs缺氧损伤,其作用机制可能与抑制miR-455表达及PI3K/Akt信号通路活化有关,抑制MEG3表达可抑制缺氧诱导的NSCs凋亡,促进细胞增殖、分化,减轻氧化应激反应,可为临床治疗提供新方向。但脑梗死等缺氧缺血性脑血管疾病的发生机制较为复杂,关于其具体作用机制仍需进一步探讨。
