柱前衍生HPLC-UV法测定氯氮平片中乳糖的含量
2021-09-18汤建新
陈 瑶 汤建新 唐 英,
1. 湖南工业大学
生命科学和化学学院
生物医用纳米材料与器件
湖南省重点实验室
湖南 株洲 412007
2. 湖南大学
化学化工学院
化学生物传感与计量学
国家重点实验室
湖南 长沙 410082
1 研究背景
氯氮平(clozapine)是常见的非典型抗精神病药物[1]。根据药物一致性评价的要求,制剂处方中原料药及各辅料的含量需与参比制剂(reference listed drug,RLD)保持一致。医药研发技术需要保密,因而医药企业只公示氯氮平参比制剂的辅料种类,并无辅料的相关用量。故利用逆向工程研究技术,测出相关辅料的含量,对氯氮平一致性评价项目的处方研究具有重要意义[2]。
乳糖是存在于大多数哺乳动物乳中的天然二糖,由半乳糖和蔗糖组成[3]。乳糖因其安全性高,广泛用于片剂和胶囊剂的填充剂或稀释剂[4]、静脉注射剂、冻干产品和婴儿食品配方中。不同粒度的乳糖具有不同的作用,如片剂湿法制粒及伴有研磨混合的过程时,宜选择细小粒度级别的乳糖,这样更易于与其他成分混合,也可更有效发挥黏合剂的作用[5]。氯氮平作为口服制剂,工艺配方中也需加入了一定比例的乳糖作为填充剂。乳糖作为氯氮平不可或缺的辅料,确定其含量,可为氯氮平片剂工艺处方的开发提供有效数据,从而加快氯氮平片剂的仿制速度。
乳糖无紫外吸收能力。目前其检测方法有碘量法[6]、滴定法[7]、酶解法[8]、比色法[9]、旋光仪法[10]、近红外光谱法[11-12]、拉曼光谱法[13],以及高效液相色谱(high-performance liquid chromatography,HPLC)与示差折光检测器[14-18]、蒸发光散射检测器[19-20]、质谱检测器[8,21]、电喷雾检测器[22]等串联的方法。其中,碘量法、酶解法、滴定法、旋光仪法,这些方法存在操作复杂、技术要求高、测试时间长、灵敏度不足、准确度较低的问题;近红外光谱法及拉曼光谱法需结合化学计量学建立定量模型,模型的建立需要大量样品和预先测定目标值,成本较高;高效液相色谱与示差折光检测器、蒸发光散射检测器、电喷雾检测器、质谱检测器等串联的方法具有灵敏度高等优点,但该类仪器设备昂贵,制约其在中小型医药企业的广泛应用。因此,发展一种成本低、快速、灵敏的乳糖检测技术具有重要意义。
1-苯基-3-甲基-5-吡唑啉酮(1-phenyl-3-methyl-5-pyrazolinone,PMP)可与乳糖发生衍生反应(见图1),其衍生产物具有较强的紫外吸收能力,从而可通过高效液相色谱-紫外检测器(ultraviolet absorption detector,UV)对乳糖进行间接检测[23-24]。目前,HPLC-UV普及率高且经济,实验操作简单,准确度高。虽然HPLC-UV广泛用于中药材、食品、生物材料中乳糖含量的检测,但其应用于仿制药业领域的报道尚少。
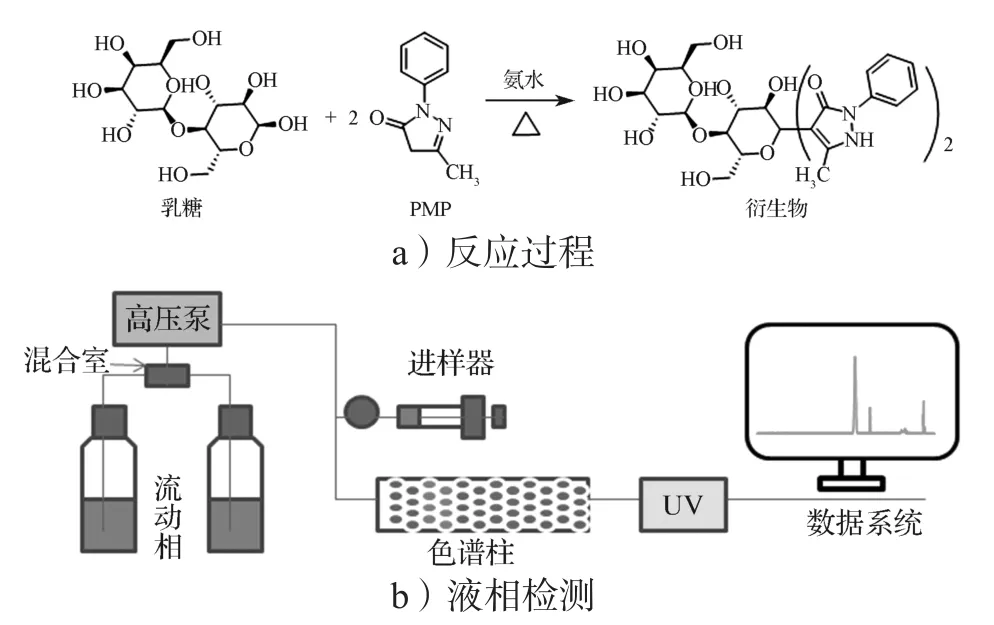
图1 乳糖和PMP反应过程及液相检测模拟Fig. 1 Lactose derivatization simulation with PMP and the detection principle of HPLC
综上所述,本研究利用PMP与乳糖进行衍生反应,再对衍生物(在245 nm处有紫外吸收)进行检测,间接定量分析氯氮平片中乳糖的含量。该方法具有简便、快速、专属性强、灵敏度高等优点,可满足氯氮平一致性评价中逆向工程研究的需求,进而加快仿制药的研发进程。
2 实验部分
2.1 主要设备与仪器、试剂与材料
1)设备与仪器
分析天平,XSE105DU型,瑞士Mettler Toledo集团;水浴锅,DF-101S型,郑州豫华仪器制造有限公司;高效液相色谱仪,Waters e2695-2998型,美国Waters公司;色谱工作站,Empower 3型,美国Waters公司;纯水仪,Exceed-Ad-32型,成都艾柯分析设备有限公司;超声清洗仪,SB-5200D型,宁波新芝生物科技股份有限公司;pH计,PHS-3C型,上海仪电科学仪器股份有限公司。
2)试剂与材料
乳糖、磷酸,均为分析纯,天津科密欧化学试剂有限公司;1-苯基-3-甲基-5-吡唑啉酮、氨水,均为分析纯,国药集团化学试剂有限公司;醋酸铵,分析纯,山东西亚化学工业有限公司;甲醇、乙腈,均为色谱纯,德国默克公司;氯氮平片参比制剂,批号为T17039,规格为25 mg,诺华集团;氯氮平片仿制样品,批号为190701、190705、190708,规格为25 mg,湖南中南制药有限公司。
2.2 实验方法
2.2.1 色谱条件
采用Inertsil ODS-3色谱柱(250 mm×4.6 mm,5 μm),流动相A为0.1 mol/L醋酸铵溶液,用磷酸调节溶液pH至5.5,流动相B为乙腈,流速为1.0 mL/min,检测波长为245 nm,柱温为30℃,进样量为10 μL,按照表1进行梯度洗脱。
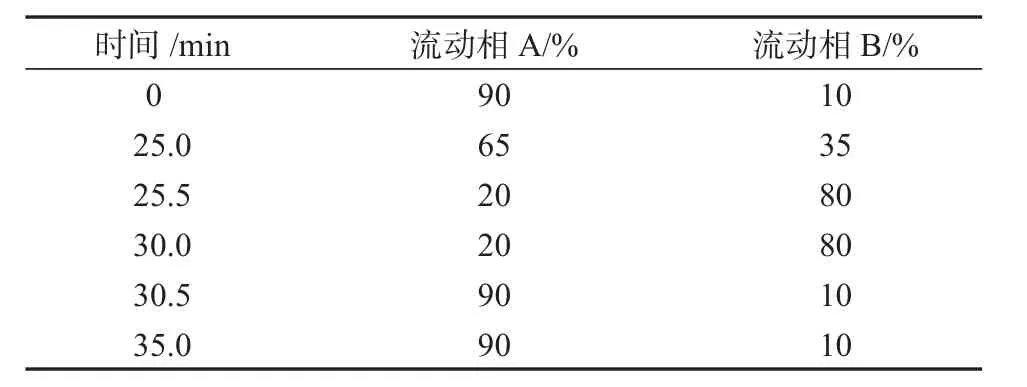
表1 流动相梯度洗脱条件Table 1 HPLC mobile phase gradient elution condition
2.2.2 溶液配制
1)空白基质:按照自研氯氮平处方混合相关辅料制得(无乳糖)。
2)衍生溶液:精确称量2.000 76 g PMP至100 mL容量瓶,再加入甲醇经超声溶解,定容,摇匀,配制成质量浓度为20.007 6 mg/mL的衍生溶液。
3)衍生空白溶液:称量12.05 mg空白基质至50 mL容量瓶,精确加入4.0 mL 20.007 6 mg/mL的PMP衍生溶液、1.0 mL纯水和0.4 mL氨水,90 ℃水浴中衍生30 min,冷却至室温,用甲醇定容,摇匀,过滤,即得。
4)对照溶液:分别精确称量220.95 mg和220.43 mg空白基质至20 mL容量瓶中,分别向容量瓶中加入200.05 mg和199.97 mg乳糖,加适量的水经超声溶解,定容后作为储备溶液;再精确移取1.0 mL储备溶液、0.4 mL氨水、4.0 mL 20.0076 mg/mL的PMP衍生溶液至50 mL容量瓶中,90 ℃水浴中衍生30 min,冷却至室温,再用甲醇定容,摇匀,过滤,即得。
5)供试品溶液:随机选取2片氯氮平片(需称量质量)至20 mL容量瓶中,加适量的水经超声至完全溶解,定容后作为储备溶液;再移取1.0 mL上述储备溶液至50 mL容量瓶中,加入0.4 mL氨水和4.0 mL 20.007 6 mg/mL 的PMP衍生溶液,90 ℃水浴中衍生30 min,冷却至室温,用甲醇定容,摇匀,过滤,即得。
3 结果与讨论
3.1 色谱条件的优化
移取1.0 mL 10.039 mg/mL的乳糖溶液和1.0 mL 2.5 mg/mL氯氮平溶液至50 mL容量瓶中,再加入4.0 mL 20.001 1 mg/mL的PMP衍生溶液和0.4 mL氨水,90 ℃水浴中加热30 min,取出冷却至室温,用甲醇定容,作为供试品溶液,用于色谱条件优化。供试品溶液经过衍生反应后,进行等度洗脱分离,结果显示衍生物保留时间约为22 min,氯氮平峰在35 min之内未出峰。为提高分析效率,将色谱条件调整为梯度洗脱,按照最佳梯度洗脱条件(见表1)进样,得到衍生目标峰、PMP和氯氮平的色谱峰如图2所示。衍生目标峰与PMP间分离度(R=7.0)良好,氯氮平峰在分析周期内得以洗脱,且分析时间适中。
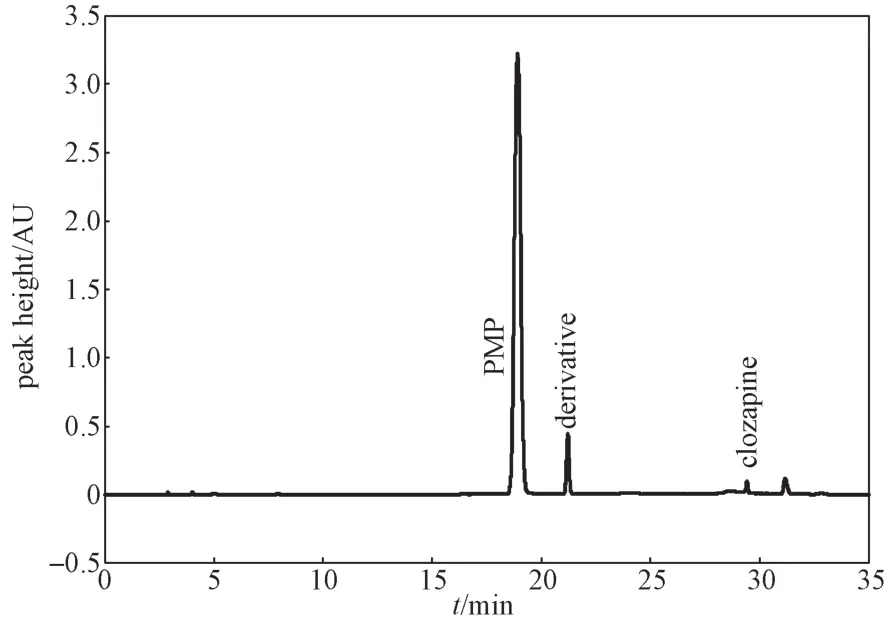
图2 供试品样品的衍生色谱图Fig. 2 Chromatogram of the test sample derivatized by PMP
3.2 衍生条件的优化
3.2.1 催化剂氨水用量的优化
移取1.0 mL 10.008 mg/mL的乳糖溶液至50 mL容量瓶,再加入4.0 mL 20.003 4 mg/mL的PMP衍生溶液,做7份平行试样,分别加入0.2, 0.4, 0.6, 0.8, 1.0,1.5, 2.0 mL的氨水,90 ℃水浴中加热30 min,取出冷却至室温,用甲醇定容。
不同体积的氨水(V1)对乳糖衍生目标峰峰面积的影响曲线如图3所示。由图可知,随着氨水用量的增加,衍生目标峰的峰面积呈现先增大后减小的趋势。当氨水体积为0.4 mL时,衍生目标峰的峰面积最大。所以氨水最优体积选择0.4 mL。
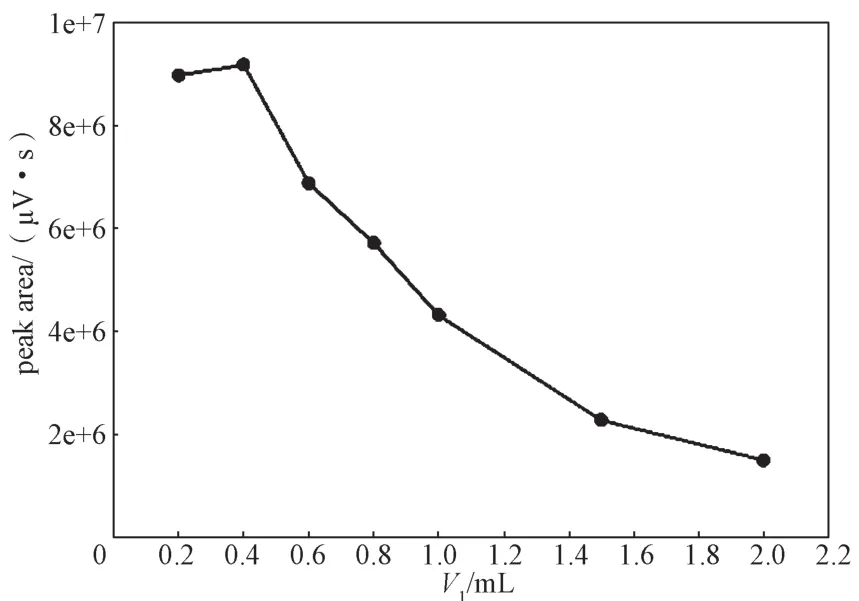
图3 乳糖衍生目标峰峰面积与氨水体积的关系图Fig. 3 The peak area of lactose’s derivative to different volume of ammonia
3.2.2 衍生试剂PMP用量的优化
移取1.0 mL 10.012 mg/mL的乳糖溶液至50 mL容量瓶,再加入0.4 mL氨水,做5份平行试样,分别加入1.0, 2.0, 3.0, 4.0, 5.0 mL质量浓度为20.000 3 mg/mL的PMP衍生溶液,90 ℃水浴中加热30 min,取出冷却至室温,用甲醇定容。
不同体积的PMP衍生溶液(V2)对乳糖衍生目标峰峰面积的影响曲线如图4所示。由图可知,随着PMP用量的增加,衍生目标峰的峰面积增加幅度不明显,呈现波动变化。但当PMP体积为4.0 mL时,衍生峰面积出现最大值,且当PMP体积为5.0 mL时,衍生峰面积明显减小。因此,最终选择PMP衍生溶液体积为4.0 mL。
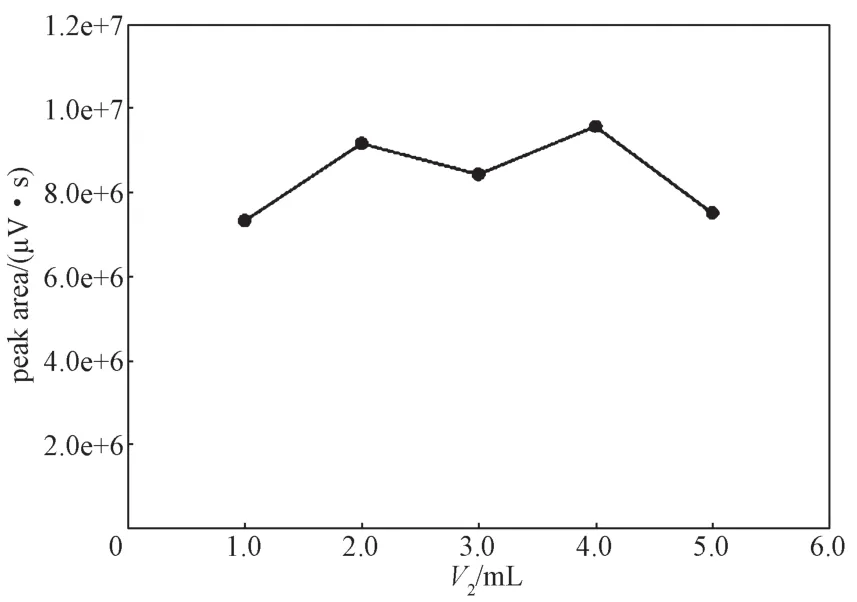
图4 乳糖衍生目标峰峰面积与PMP衍生溶液体积的关系图Fig. 4 The peak area of lactose’s derivative to different volume of PMP
3.2.3 衍生温度的优化
移取1.0 mL 10.020 mg/mL 乳糖溶液至50 mL容量瓶中,再加入0.4 mL氨水和4.0 mL质量浓度为20.001 7 mg/mL的PMP衍生溶液,做5份平行试样,分别放置于50, 60, 70, 80, 90 ℃水浴中加热30 min,取出冷却至室温后,用甲醇定容。
不同衍生温度对乳糖衍生目标峰峰面积的影响曲线如图5所示。

图5 乳糖衍生目标峰峰面积与衍生温度的关系图Fig. 5 The peak area of lactose’s derivative to different temperatures
由图5可知,随着衍生温度的增加,衍生目标峰的峰面积不断增加;当水浴温度达到80 ℃时,衍生目标峰的峰面积达到平台期,说明该温度下衍生反应基本完成。但为使衍生反应彻底并降低结果波动性的影响,最终选择90 ℃作为衍生温度。
3.2.4 衍生时间的优化
移取1.0 mL质量浓度为10.000 7 mg/mL的乳糖溶液至50 mL容量瓶中,再加入0.4 mL氨水和4.0 mL质量浓度为20.002 mg/mL的PMP衍生溶液,做7份平行试样,90 ℃水浴中分别加热15, 20, 25, 30,35, 40, 50 min,取出冷却至室温,用甲醇定容。
不同衍生时间对乳糖衍生目标峰峰面积的影响曲线如图6所示。由图可知,在30 min前,衍生目标峰的峰面积不断增大;在30~40 min,峰面积呈现减小趋势;过了40 min,峰面积又呈现增大趋势。在50 min内,衍生目标峰的峰面积在30 min时达到最大值。故最优的衍生时间为30 min。
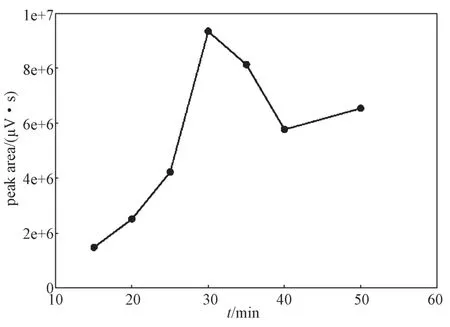
图6 乳糖衍生目标峰峰面积与衍生时间的关系图Fig. 6 The peak area of lactose’s derivative to different derivation time
3.3 实验方法的性能验证
3.3.1 系统精密度
按照2.2.2的方法配制对照溶液,重复进样对照溶液6针,计算6针衍生目标物的峰面积和保留时间的相对标准偏差(relative standard deviation,RSD)分别为0.85%和0.08%,由此说明进样系统的精密度良好。
3.3.2 稳定性
按照方法2.2.2配制对照溶液和供试品溶液,将对照溶液和样品溶液在室温分别放置0, 2, 4, 8, 12, 24,36, 48 h后进样,计算衍生目标峰峰面积的相对标准偏差分别为0.38%和0.48%。该结果表明对照溶液和供试品溶液在室温48 h内稳定,后续批量制备溶液和进样可在室温48 h内完成。
3.3.3 专属性
图7为衍生空白溶液、对照溶液、氯氮平样品溶液的色谱图。由图可以看出,PMP、乳糖衍生目标物、氯氮平的保留时间分别为19.348 , 22.243, 29.484 min,各化合物之间分离度良好,且完全不干扰衍生目标峰,由此说明该方法专属性良好。
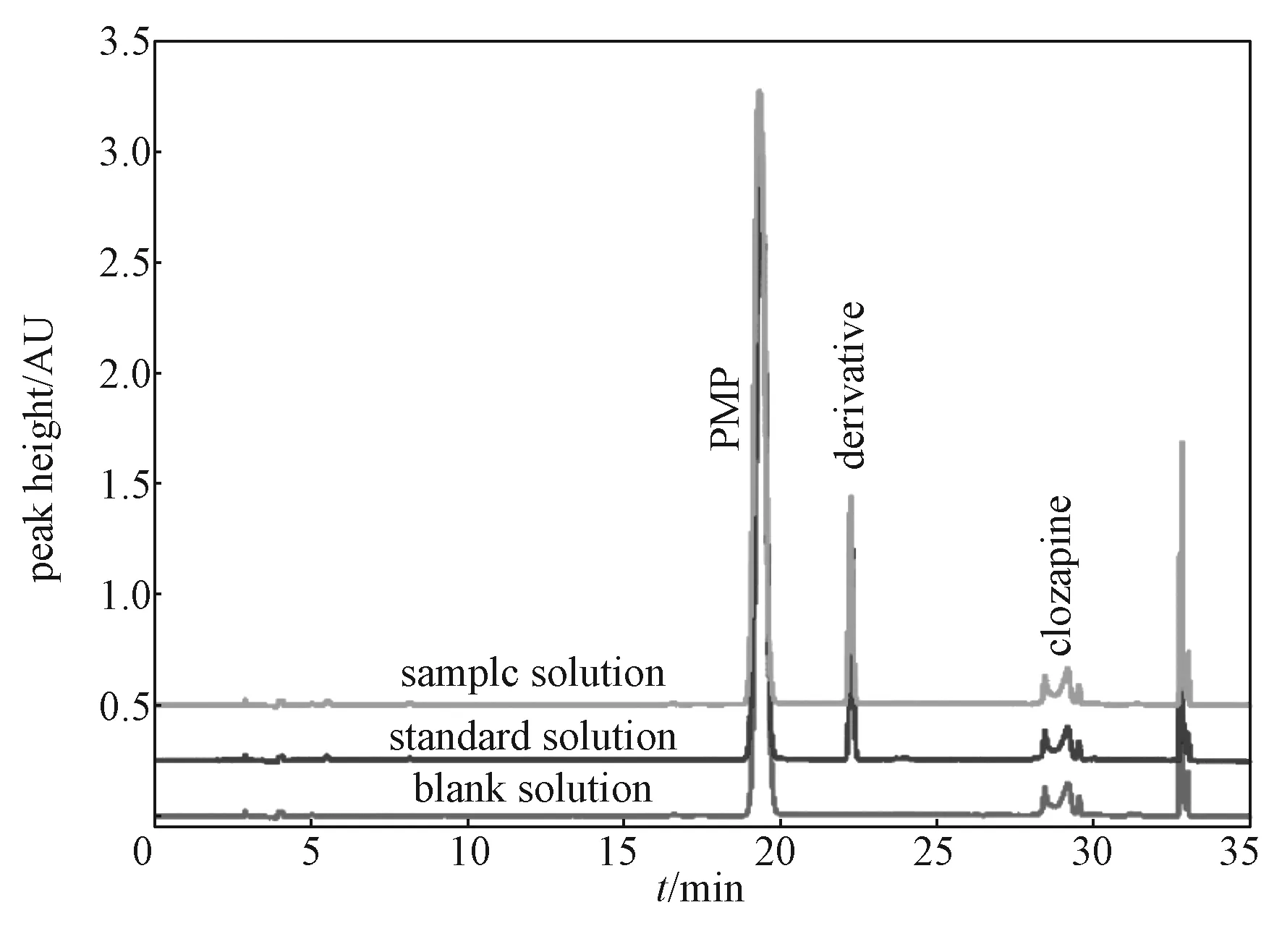
图7 衍生空白溶液、对照溶液、氯氮平样品溶液的色谱图Fig. 7 Chromatogram of derivative blank solution,control solution and clozapine sample solution
3.3.4 线性范围和定量限
以衍生目标物的峰面积为纵坐标,乳糖的质量浓度c(mg/mL)为横坐标,建立标准加入法的标准曲线(如图8),其线性回归方程为:y=41 856 509.560 5x+36 592.315 8,相关系数R2为0.999 4。实验结果表明,在0.060 4~0.399 7 mg/mL范围内,衍生目标物的峰面积与乳糖质量浓度呈良好的线性关系。再将乳糖储备溶液逐步稀释后进行衍生,直至信噪比(S/N)在10~20之间,得到乳糖定量限为0.32 μg/mL。
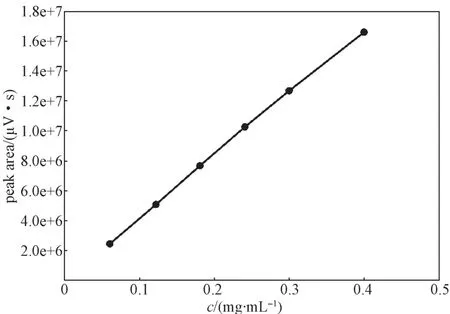
图8 乳糖衍生目标物的峰面积与浓度关系图Fig. 8 The peak area of lactose’s derivative to different concentration
同时,将该方法与其他一些报道的乳糖检测方法进行比较(见表2)。本方法不需要昂贵的设备仪器、实验操作简单、具有较宽的线性响应范围、准确度高,能满足氯氮平片中乳糖的检测要求。
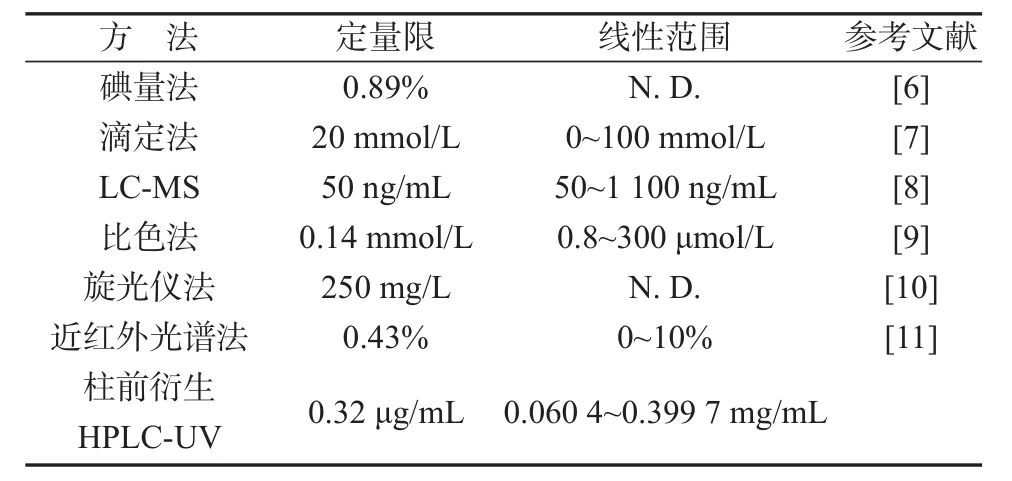
表2 不同方法检测乳糖的检出限和线性范围Table 2 Detection limits and linear ranges of different methods for lactose detection
3.3.5 耐用性
将对照溶液和供试品溶液,分别按照流速(1.0±0.1)mL/min、柱温(30±5)℃、检测波长(245±2)nm改变色谱参数及换用不同品牌色谱柱进样对照溶液和供试品溶液,计算的乳糖含量相对标准偏差为1.04%,且目标峰无相关干扰,由此说明本方法的耐用性及抗干扰性良好。
3.3.6 回收率
取9份供试品溶液,按照高、中、低3个水平添加乳糖作为加标溶液,考察本方法的回收率,结果如表3所示。
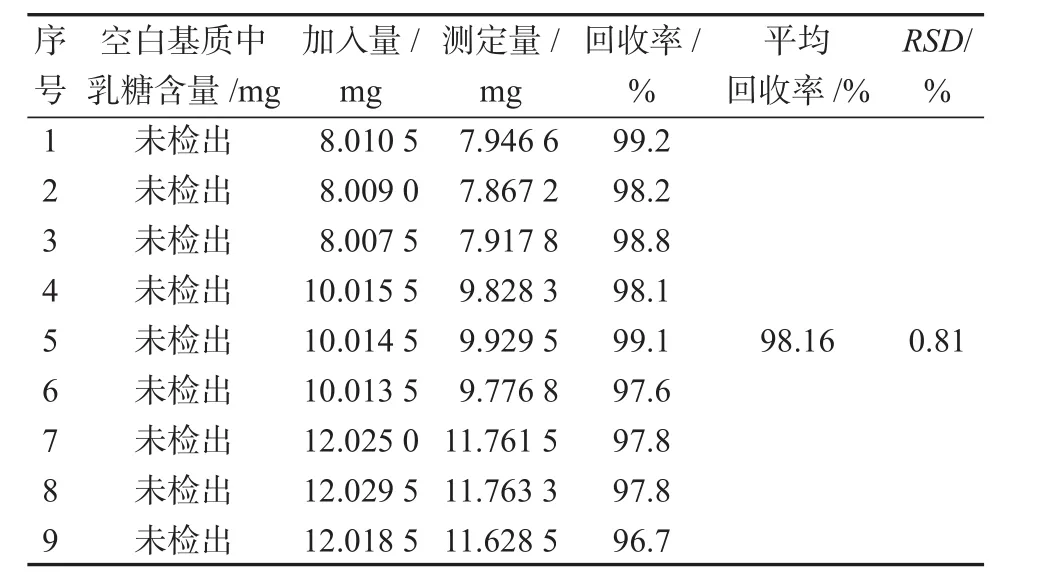
表3 乳糖在空白基质中的加标回收率(n=9)Table 3 Spiked recoveries of hydrazine in blank substrate (n=9)
由表3可知溶液,其回收率在96.7%~99.2%,平均回收率为98.16%,RSD为0.81%。各个浓度水平的回收率结果均较好,且相对标准偏差较小,由此说明本方法检测结果的准确度高,重复性较好。
3.4 样品检测
按照处方,仿制3批(批号为190701, 190705,190708)乳糖含量已知的氯氮平片样品和参比制剂(批号为T17039)样品,按照上述方法衍生后测定乳糖含量,经过换算得出4个批次乳糖含量及偏差如表4所示。
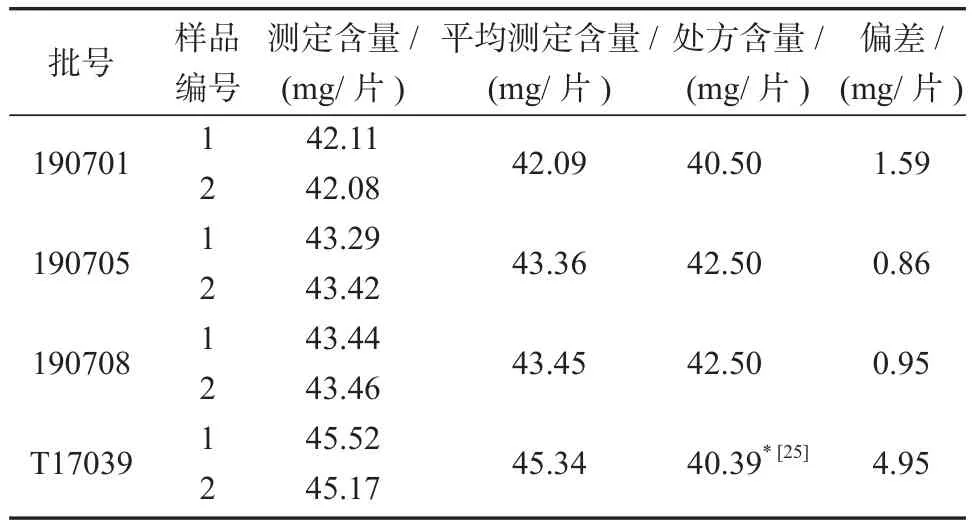
表4 自制处方和参比制剂中乳糖含量的测定结果Table 4 Determination results of lactose content in homemade prescription and reference preparation
从表4数据可知,本法测定的自制处方中乳糖含量实测值分别为42.09, 43.36, 43.45 mg/片,与处方含量的偏差均小于2.0 mg/片。参比制剂测得乳糖的含量为45.34 mg/片,但其对照含量40.39 mg/片是根据文献[25]报道的,该文献利用的是HPLC结合示差折光检测器检测氯氮平中乳糖的含量。因为检测器的不同、参比制剂的批间差异、方法测定误差等原因,综合导致参比制剂检测结果中乳糖含量检测偏差为4.95 mg/片。本文自制处方的检测结果与实际处方含量相差较小,均小于2.0 mg/片,而文献[25]检测方法中自制检测结果与其实际含量偏差在2.91 mg/片,较本法结果误差偏大。且文献中的检测方法需要使用氨基柱这种特殊类型的色谱柱,其氨基柱的键合官能团氨丙基容易水解,在进行样品检测时,流动相中水的比例较高,易引起氨丙基的水解,从而影响色谱柱的使用寿命。官能团氨丙基带有负电荷,在分析酸性物质(如糖类)时,容易与带质子的酸性物质发生反应,使得氨基柱的柱效降低,影响检测的结果,故需要对氨基柱进行维护。本方法中使用的色谱柱填料为常规的C18填料,分析方法中水相比例可放宽调整,也不会因色谱柱维护不当影响结果检测。因此,利用本实验方法测定的参比制剂数值,可以加快一致性评价项目的处方研究,缩短研发周期和降低研发成本。
4 结论
药物一致性评价要求仿制药与参比制剂在剂型、处方、规格及给药途径等各方面保持一致。仿制药同参比制剂越相似,生物等效性成功的可能性就越大。因此,本研究提出了柱前衍生HPLC-UV法,测定氯氮平片中乳糖含量,即用PMP作为衍生试剂与乳糖衍生后,用HPLC-UV法间接测定氯氮平中乳糖含量。该方法各项参数指标符合中华人民共和国药典的要求,具有经济、简便、专属性强及准确度高等优点,可用于氯氮平片中乳糖的含量测定,进而可推广到其他仿制药中乳糖的逆向研究中。
