TNF-α对类风湿关节炎滑膜成纤维细胞自噬和增殖的影响及作用机制研究
2021-09-16汪吉烽王维维吴颖慧罗晨栩窦瑞玲高玲王国芬莫选荣罗心静
汪吉烽 王维维 吴颖慧 罗晨栩 窦瑞玲 高玲 王国芬 莫选荣 罗心静
类风湿性关节炎(rheumatoid arthritis,RA)是一种慢性炎症性自身免疫疾病,其主要的病理特征是由于关节滑膜组织增生和炎症而导致的软骨及骨的损伤。RA患者关节组织中的滑膜成纤维细胞(rheumatoid arthritis synovial fibroblasts,RASF)是滑膜炎症的主要效应细胞,其过度增殖造成的细胞数量异常增多是滑膜增生和骨质破坏的重要机制[1]。近年的研究表明,TNF-α、IL-1等炎症因子诱导的RASF自噬上调是其凋亡抵抗和异常增殖的重要原因之一,并与RA滑膜增生密切相关[2]。因此调控RASF自噬所导致的滑膜增生已成为治疗RA的有效策略[3]。NF-κB是在RA发病中起重要作用的炎症信号通路之一,其在RA滑膜组织中高度活化,参与炎症因子诱导的滑膜炎症反应[4]。但其对炎症因子诱导的RASF自噬的影响尚不太清楚。本研究拟以人类RASF细胞株MH7A作为载体,观察TNF-α对RASF自噬和增殖的影响,并进一步通过抑制NF-κB通路来探讨这一影响的作用机制。
1 材料和方法
1.1 主要材料和试剂 MH7A购自广州吉妮欧公司;改良杜氏伊格尔培养基(DMEM)、FBS均购自美国Gibco公司;Bay11-7082购自美国MedChemExpress公司;总RNA分离试剂盒(Trizol)购自美国Applied Biosystems公司;甘油醛-3-磷酸脱氢酶(GAPDH)多克隆抗体购自美国GOOD HERE公司,辣根过氧化物酶(HRP)标记的山羊抗兔IgG购自美国Santa Cruz公司;重组人TNF-α购自美国Novoprotein公司;噻唑蓝法(MTT)试剂盒、荧光二抗、4,6-联脒-2-苯基吲哚(DAPI)、十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)凝胶配制试剂盒均购自南通碧云天公司;唯Bcl-2同源-3域蛋白-1(Beclin-1)兔单克隆抗体、磷酸化 NF-κB-p65(P-p65)兔单抗均购自美国Cell Signaling Technology公司;自噬相关基因微管相关蛋白1轻链3-Ⅱ/自噬相关基因微管相关蛋白1轻链3-Ⅰ(LC3-Ⅱ/LC3-Ⅰ)单抗、腺病毒-绿色荧光蛋白(Ad-GFP)-LC3-Ⅱ试剂、PCR试剂盒均购自美国Thermo公司。
1.2 方法
1.2.1 RASF传代和培养 将MH7A用含10%FBS的DMEM培养液培养,每隔3~4 d换液1次,当细胞生长至融合度80%时用0.25%胰蛋白酶消化,按1∶3比例分瓶传代并继续培养。
1.2.2 实验分组 在一阶段实验中,根据5 ng/ml TNF-α 处理时间不同,将细胞分为 6 组,即 0、3、6、12、24 及48 h组。在二阶段实验中,根据加入的药物不同,将细胞分为4组,即5 μg/ml Bay11-7082预处理的Bay组,5 ng/ml TNF-α 预处理的 TNF-α 组,5 μg/ml Bay11-7082预处理2 h后再用5 ng/ml TNF-α预处理的Bay+TNF-α组,细胞不作处理的正常对照(Ctrl)组。
1.2.3 细胞增殖活性检测 采用MTT法。将上述各组预处理细胞用胰酶消化并等量接种于96孔板,生长至一定密度加入20 μl MTT溶液,继续培养4 h;终止培养后,小心吸去培养液,每孔加入150 μl二甲基亚砜,摇床15 min,在492 nm波长处用酶标仪测量吸光度(OD)值。细胞相对活性=实验组OD值/Ctrl组OD值×100%。
1.2.4 细胞中自噬小体形成情况检测 共聚焦显微镜下采用GFP-LC3-Ⅱ来示踪自噬形成。将上述各组预处理细胞用胰酶消化并接种于共聚焦培养皿,生长至一定密度后加入7.5 μl Ad-GFP-LC3-Ⅱ,继续培育24 h;PBS漂洗,固定液静置15 min;再经PBS漂洗后每孔加入80 μl的DAPI,避光摇床15 min;吸去染色液,PBS漂洗3遍;在共聚焦显微镜下观察外源性GFP-LC3-Ⅱ在细胞定位,分析细胞中自噬小体的形成情况。
1.2.5 P-p65、LC3-Ⅱ/LC3-Ⅰ和 Beclin-1蛋白表达水平检测 采用Western blot法。将上述各组预处理细胞用PBS缓冲液漂洗3次,加入蛋白裂解液冰上混匀裂解30 min,离心取上清液,采用BCA法测出蛋白浓度;将 20 μg蛋白样品与6×SDS加样缓冲液混合上样;样品经10%SDS-PAGE电泳分离后,经电转移法转移到PVDF膜上,室温下封闭4 h;分别加入GAPDH、P-p65、LC3-Ⅱ/LC3-Ⅰ和Beclin-1兔多克隆抗体,孵育过夜;隔日漂洗后,加入HRP标记的羊抗兔IgG,反应1 h;加入ECL试剂,曝光成像并灰度扫描分析。GAPDH为内参照物。
1.2.6 Beclin-1 mRNA的表达水平检测 采用real-time PCR法。用Trizol法提取RNA,将提取后的RNA经逆转录制备 cDNA,反应体系如下:1 μg RNA、40 U/μl莫罗尼小鼠白血病病毒逆转录酶、100 nmol/L多聚胸腺嘧啶、1 mmol/L脱氧核糖核苷三磷酸混合物、0.1 mol/二硫苏糖醇、40 U/μl RNA酶抑制物、加焦碳酸二乙酯去离子水至总体积为20 μl。反应条件如下:42℃60 min,70℃10 min。将生成的cDNA进行PCR扩增,反应体系如下:0.4 μmol/L cDNA、1 μl正、反义链 12.5 μl SYBR Green酶、加去离子水至总体积20 μl。PCR反应程序如下:95℃5 min预变性,然后按95℃15 s,60℃1 min共40个循环,最后72℃7 min延伸。计算模板与对照样本之间的2-ΔΔCt值差。
1.2.7 P-p65在细胞中表达及定位检测 采用细胞免疫荧光法。将将上述各组预处理细胞用胰酶消化并等量移至细胞爬片;入封固液1 h。加入NF-κB p65,4℃过夜,隔日用PBS清洗3次,加入羊抗兔IgG室温下摇床2 h。用DAPI(1∶1 000)染核(全程避光),用PBS漂洗3次,在新的载玻片上滴加淬灭液,置于荧光倒置显微镜下观察。

2 结果
2.1 不同时间6组细胞相对活性比较 与0 h组相比,12、24、48 h组细胞相对活性均明显升高(均P<0.05),见表 1。

表1 不同时间6组细胞相对活性比较(%)
2.2 不同时间6组自噬小体形成情况比较 GFPLC3-Ⅱ腺病毒转染细胞后,与0 h组相比,TNF-α刺激滑膜细胞后自噬小体形成明显增多,其中24 h组最为明显,见图1(插页)。
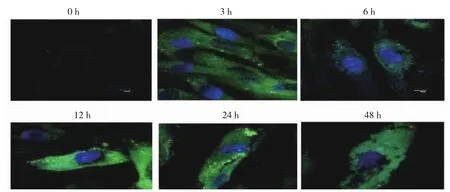
图1 不同时间6组细胞TNF-α刺激后自噬小体形成共聚集显微镜下所见(×200)
2.3 不同时间6组细胞LC3-Ⅱ/LC3-Ⅰ及Beclin-1蛋白表达水平比较 与0 h组相比,3、6、12 h组LC3-Ⅱ/LC3-Ⅰ蛋白表达水平均明显升高,差异均有统计学意义(均 P<0.05),6、12、24、48 h 组 Beclin-1 蛋白表达水平均明显升高,差异均有统计学意义(均P<0.05),见图2、表2。

表2 不同时间6组细胞LC3-Ⅱ/LC3-Ⅰ、Beclin-1蛋白表达水平比较
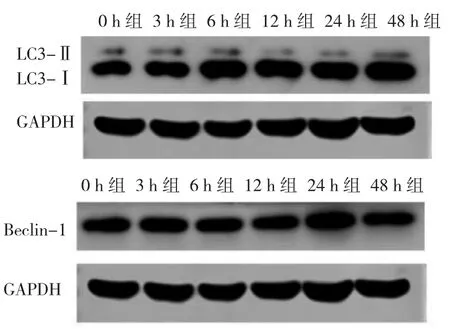
图2 不同时间6组细胞LC3-Ⅱ/LC3-Ⅰ及Beclin-1蛋白表达的电泳图(LC3-Ⅱ/LC3-Ⅰ为自噬相关基因微管相关蛋白1轻链3-Ⅱ/自噬相关基因微管相关蛋白1轻链3-Ⅰ;Beclin-1为唯Bcl-2同源-3域蛋白-1)
2.4 不同时间6组细胞Beclin-1 mRNA表达水平比较 与 0 h 组相比,6、12、24、48 h 组 Beclin-1 mRNA表达水平均明显升高,差异均有统计学意义(均P<0.05),见表3。

表3 不同时间6组细胞Beclin-1 mRNA表达水平比较
2.5 不同药物预处理4组细胞P-p65蛋白表达及染核情况比较 与TNF-α组相比,Bay+TNF-α组细胞内P-p65蛋白表达及染核明显减少,见图3(插页)。与Ctrl组比较,Bay组细胞P-p65蛋白的表达水平明显下降,与TNF-α组比较,Bay+TNF-α组细胞P-p65蛋白的表达水平明显下降,差异均有统计学意义(均P<0.05),见图4。

图3 不同药物预处理4组细胞NF-κB染核情况荧光显微镜下所见(P-p65为磷酸化NF-κB-p65;DAPI染色,×200)

图4 不同药物预处理4组细胞P-p65蛋白表达的电泳图(P-p65为磷酸化 NF-κB-p65)
2.6 不同药物预处理4组细胞相对活性比较 与Ctrl组比较,Bay组细胞相对活性明显降低,差异有统计学意义(P<0.05);与 TNF-α 组比较,Bay+TNF-α 组细胞相对活性明显降低,差异有统计学意义(P<0.05),见表4。

表4 不同药物预处理4组细胞相对活性比较(%)
2.7 不同药物预处理4组细胞自噬小体形成情况比较 与TNF-α组相比,Bay+TNF-α组细胞自噬小体明显减少,见图5(插页)。
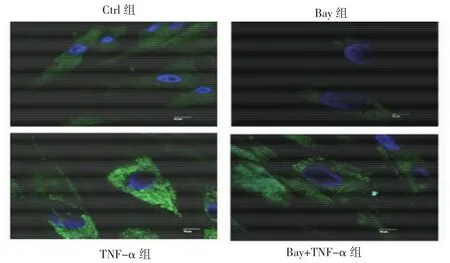
图5 不同药物预处理4组细胞自噬小体形成共聚集显微镜下所见(×200)
2.8 不同药物预处理4组细胞LC3-Ⅱ/LC3-Ⅰ及Beclin-1蛋白表达水平比较 与Ctrl组比较,Bay组LC3-Ⅱ/LC3-Ⅰ、Beclin-1蛋白表达水平均明显下降,差异均有统计学意义(均P<0.05);与TNF-α组比较,Bay+TNF-α组LC3-Ⅱ/LC3-Ⅰ、Beclin-1蛋白表达水平均明显下降,差异均有统计学意义(均P<0.05),见图6、表5。

图6 不同药物预处理4组细胞LC3-Ⅱ/LC3-Ⅰ、Beclin-1蛋白表达的电泳图(LC3-Ⅱ/LC3-Ⅰ为自噬相关基因微管相关蛋白1轻链3-Ⅱ/自噬相关基因微管相关蛋白1轻链3-Ⅰ;Beclin-1为唯Bcl-2同源-3域蛋白-1)

表5 不同药物预处理4组细胞LC3-Ⅱ/LC3-Ⅰ、Beclin-1蛋白表达水平比较
2.9 不同药物预处理4组Beclin-1 mRNA表达水平比较 与Ctrl组比较,Bay组Beclin-1 mRNA表达水平明显下降,差异有统计学意义(P<0.05);与TNF-α组比较,Bay+TNF-α组Beclin-1 mRNA表达水平明显下降,差异有统计学意义(P<0.05),见表6。

表6 不同药物预处理4组细胞Beclin-1 mRNA表达水平比较
3 讨论
RA病变的一个重要特征就是由于滑膜组织的增生所导致的关节软骨及骨的侵蚀破坏。研究表明RASF是引起关节滑膜组织增生的关键效应细胞。RASF与正常滑膜细胞不同,在炎症因子(如TNF-α、IL-1)等刺激下,其细胞表型及生物学行为均发生显著变化,具有“肿瘤样”生长和侵袭特性,能逃避正常的生长调控机制而出现大量增殖,并合成和分泌多种炎性细胞因子、趋化因子和基质金属蛋白酶,导致慢性滑膜炎,形成血管翳并侵蚀软骨和软骨下骨[5]。如果能够有效抑制RASF活化和异常增殖,将能阻断滑膜增生和骨质侵蚀,从而使RA的病变得到有效的控制。然而,RASF异常增殖的机制十分复杂,目前尚不明确。
近年来研究结果表明,RASF自噬参与RA滑膜增生病变过程[6-8]。RASF自噬上调是RASF凋亡抵抗及过度增殖的重要机制,在RA滑膜增生和关节破坏中起着重要作用。某些在RA病变中起重要作用的炎症因子(如 TNF-α、IL-1、IL-6、IL-17等)在 RASF自噬产生过程中发挥着重要作用[9]。如TNF-α不仅能诱导RASF凋亡抵抗,还能诱导RASF的自噬,故其已被广泛用作RA的治疗靶标[10-11]。本研究一阶段实验结果证实,RASF受TNF-α刺激后细胞增殖活性显著增加,同时自噬细胞数及自噬标志性蛋白LC3-Ⅱ/LC3-Ⅰ水平升高,提示TNF-α能诱导RASF自噬,并且与细胞增殖相关。Beclin-1是酵母自噬相关基因Atg6的同源物,属于Bcl-2抗癌基因家族,其不但参与细胞凋亡调节,而且参与细胞自噬的调控。本研究发现,TNF-α刺激RASF后Beclin-1 mRNA和蛋白表达水平均升高,表明Beclin-1参与TNF-α诱导RASF自噬过程,可能与RA病变有关。
目前NF-κB通路是与RA发病关系最为明确的信号转导通路。研究表明,NF-κB介导RASF活化及多种炎症细胞因子的生成调节[12]。NF-κB也是RASF存活的关键调节因子,参与RA滑膜增生的发病机制[4]。本研究二阶段实验中,采用NF-κB通路抑制剂BAY11-7082来抑制NF-κB通路活化,观察TNF-α刺激下的RASF增殖和自噬的变化,结果显示,抑制NF-κB通路活化能显著降低基础状态和TNF-α刺激下的RASF增殖和自噬水平,同时Beclin-1基因表达显著降低。以上研究提示,NF-κB通路活化介导了TNF-α诱导的RASF自噬和增殖活性,并且可能与其上调Beclin-1表达有关。有研究报道抑制NF-κB活化能明显抑制RASF增殖并促进凋亡[4]。而NF-κB通路是否通过上调RASF自噬促进其凋亡抵抗及其过度增殖,尚需要进一步研究。
综上所述,本实验表明TNF-α能诱导RASF自噬和增殖活性,其机制与其激活NF-κB通路活化有关。因此,靶向NF-κB介导的RASF自噬的调控,可能成为有效防治RA的新靶点。
