HPLC-DAD鉴别皂角刺中掺入野皂角刺的方法研究
2021-09-15刘小燕夏巧红宋平顺张义福贺军权
刘小燕,夏巧红,宋平顺,张义福,贺军权,倪 琳*
(1. 甘肃省药品检验研究院 国家中药材及饮片质量控制重点实验室,甘肃 兰州 730070;2. 甘肃中医药大学 药学院,甘肃 兰州 730030)
《中国药典》2020年版(一部)收录的皂角刺为豆科植物皂荚Gleditsia sinensisLam.的干燥棘刺[1]。皂荚系豆科云实亚科皂荚属植物[2],是我国特有的乡土树种[3]。皂角刺含有多酚、黄酮、香豆素、三萜皂苷、三萜、内脂、甾醇等成分[4-5],具有消肿托毒、排脓、杀虫之功效,临床用于痈疽初起或脓成不溃,外用于疥癣麻风[1]等症的治疗,具有抗炎[6]、抑菌[7]、免疫调节、抗肿瘤[4-5]等药理活性。
近年来,随着医疗市场改革,中药饮片以次充好、以假乱真的现象层出不穷,严重影响了用药安全和临床疗效[8]。一些掺假中药材或饮片与正品极为相似,极易混淆。因此,对中药真伪的鉴别显得尤为重要。皂角刺常混伪品以野皂角刺较为多见,野皂角刺为豆科植物野皂荚Gleditsia heterophyllaBunge.带枝条的棘刺,两者属于同科同属近缘种植物,外观十分相似[9-11],传统的性状、显微、理化鉴别方法不能分辨皂角刺正品与伪品。王丽君等[12]建立了皂角刺中野皂角刺掺伪量的近红外光谱预测模型,但模型预测的准确性直接受到药材产地及炮制方法等的限制。本研究基于特征图谱的思路,参考相关文献资料,并以阿魏酸对照品作为内标物,对野皂角刺中的特有峰进行定性分析,拟建立HPLC法鉴别正品皂角刺与伪品野皂角刺。为皂角刺中掺伪野皂角刺情况评价提供依据,也可为市场其它药材掺伪掺假分析提供参考。
1 仪器与试药
1.1 仪器
Agilent 1260型高效液相色谱仪(美国安捷伦科技公司);Waters ARC型高效液相色谱仪(美国沃特世公司);Mettler MS205DU型电子天平(瑞士梅特勒-托利多公司)。
1.2 试药
阿魏酸对照品(批号:110773-201614,中国食品药品检定研究院,纯度:99.0%);皂角刺对照药材(批号:121210-201504,中国食品药品检定研究院);甲醇、乙腈(HPLC级,德国Merck公司),甲酸(优级纯,阿拉丁试剂有限公司),水为纯化水。
共收集到皂角刺正品药材及饮片7批,其基原为豆科植物皂荚Gleditsia sinensisLam.;收集到伪品野皂角刺6批,其基原为豆科植物野皂荚Gleditsia heterophyllaBunge.。样品信息见表1。皂角刺与野皂角刺均由甘肃省药品检验研究院宋平顺主任药师收集并鉴定。同时收集皂角刺药材及饮片样品26批,编号为Yp01-Yp26。
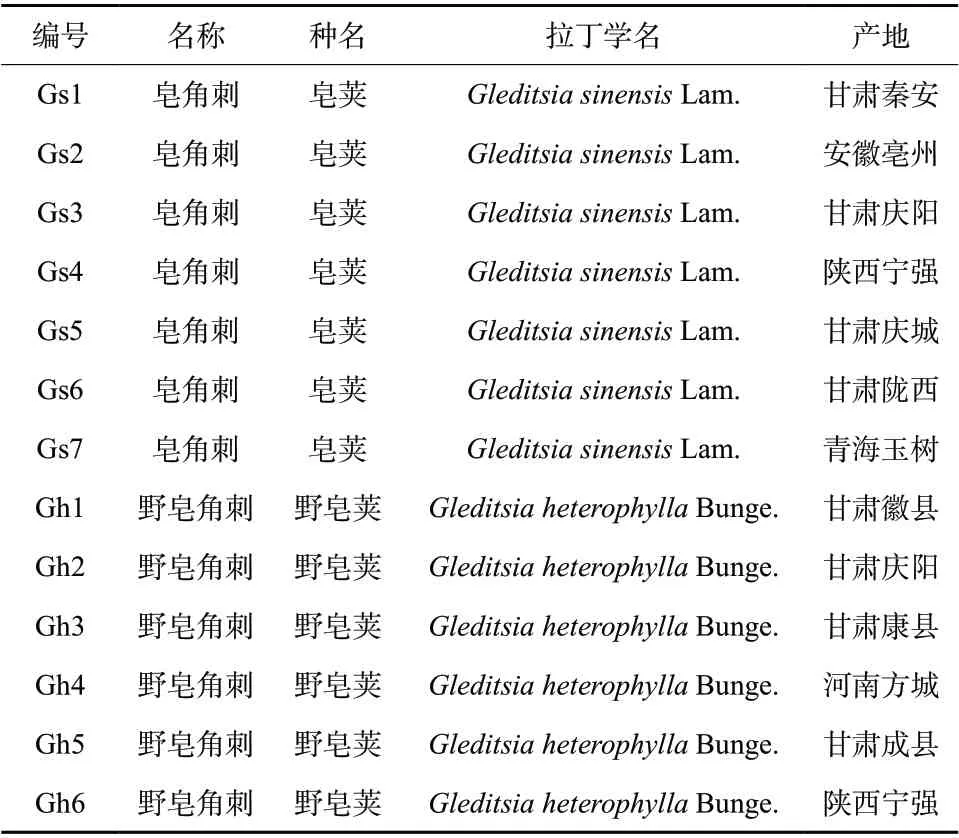
表1 皂角刺和野皂角刺的相关样品信息Tab. 1 Sample information of Gleditsia sinensisLam. and Gleditsia heterophylla Bunge.
2 方法与结果
2.1 色谱条件
ODS 色谱柱(型号:5TC-C18;填料:十八烷基硅烷键合硅胶; 4.6 mm × 250 mm,5 µm;美国安捷伦科技公司)。流动相A为0.1%甲酸,流动相B为乙腈,按表2进行梯度洗脱,柱温为30℃;检测波长为305 nm;流速为0.9 mL/min;进样量为10 µL。
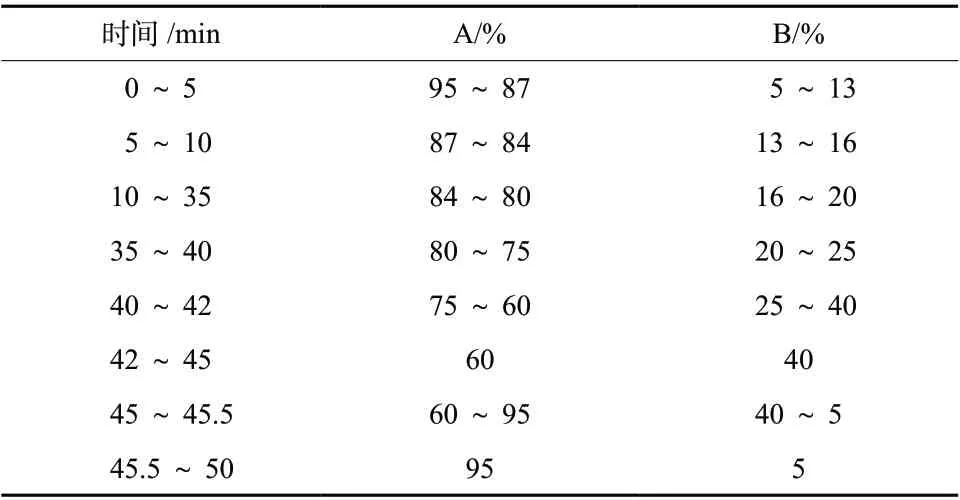
表2 流动相梯度洗脱程序Tab. 2 The gradient elution program of mobile phase
2.2 溶液的制备
2.2.1 内标溶液的制备 取阿魏酸对照品,精密称定,加甲醇制成每1 mL含0.1 mg的溶液,即得。
2.2.2 阴性对照溶液的制备 取皂角刺对照药材,过三号筛,取约0.3 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,密塞,称定重量,超声处理(功率:400 W,频率:40 kHz)30 min,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液5 mL,内标溶液1 mL,置同一10 mL容量瓶中,加甲醇稀释至刻度,摇匀,作为阴性对照溶液。
2.2.3 阳性对照溶液的制备 取野皂角刺粉末,过三号筛,自“取约0.3g”起,同“2.2.2”法制备。典型色谱图见图1。

图1 阳性对照溶液(a)、阴性对照溶液(b)及内标溶液(c)典型色谱图Fig.1 The typical chromatogram of positive control solution(a),negative control solution(b) and internal standard solution(c)
2.2.4 供试品溶液的制备 取本品粉末,过三号筛,自“取约0.3 g”起,同“2.2.2”法制备。
2.2.5 掺伪样品的制备 分别取皂角刺Gs5及野皂角刺Gh1粉末,过三号筛,按重量百分比(W/W)进行混合,同“2.2.4”法配制成皂角刺中掺入伪品野皂角刺5%、10%、20%、30%、40%、50%、60%、70%不同比例的溶液。
2.3 方法学验证
2.3.1 专属性试验 取皂角刺及伪品野皂角刺,按上述“2.2.4”法制备供试品溶液,进样。结果6批伪品野皂角刺中均检出一特有峰,与阿魏酸的相对保留时间(relative retention time,RRT)为1.43,7批正品皂角刺在与阿魏酸RRT相同的位置均未出现色谱峰,见图2;该特有峰经HPLC-DAD扫描3D图提取的光谱图,见图3。表明该方法专属性良好。

图2 皂角刺(A)与野皂角刺(B)样品色谱图Fig.2 The chromatogram of Gleditsia sinensisLam.(A)and Gleditsia heterophylla Bunge.(B)
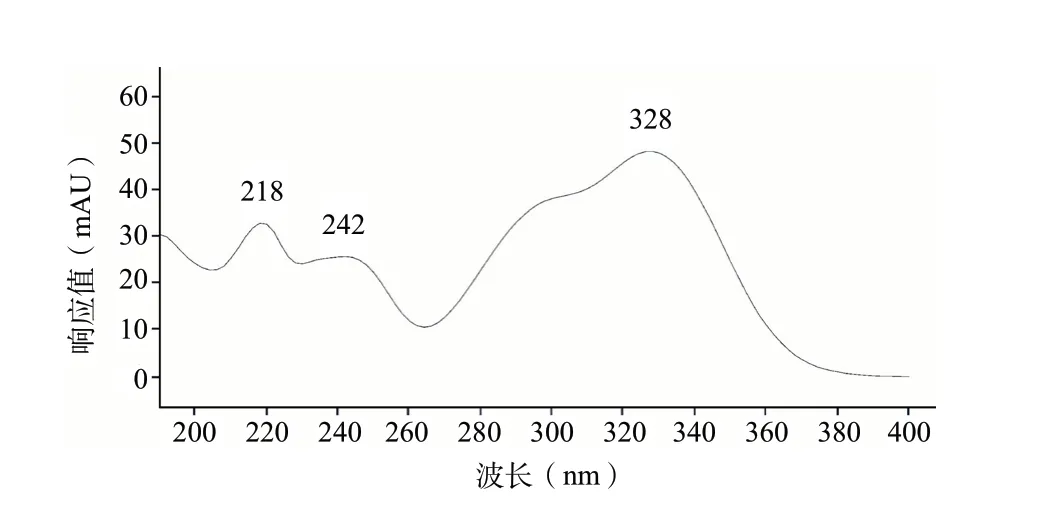
图3 3D扫描特有峰典型光谱图Fig.3 The typical spectra of peak S scanned by 3D
2.3.2 线性关系 取“2.2.5”项下掺伪样品,考察野皂角刺掺伪比例与特有峰峰面积是否呈线性关系。以掺伪比例为横坐标,以特有峰/阿魏酸峰面积比为纵坐标,绘制标准曲线,求得回归方程为
Y= 0.760 1X+ 2.386,r= 0.995 3。
2.3.3 精密度 取掺伪比例为30%的供试品溶液,重复进样6次,测得特有峰的峰面积RSD为0.4%(n= 6),与阿魏酸RRT的RSD为0.1%(n= 6),表明该方法精密度良好。
2.3.4 重复性 取掺伪比例为30%供试品溶液6份,进样。掺伪比例分别为29.8%、30.1%、30.5%、30.3%、29.8%、29.6%时,特有峰的峰面积为118.28、123.18、131.70、123.86、126.18、117.65。
RRT的RSD为0.2% (n= 6),表明该方法重复性良好。
2.3.5 稳定性 取掺伪比例为30%供试品溶液,分别于0、2、4、6、8、10、12、24 h进样,测得特有峰峰面积RSD 为0.8% (n= 8),RRT的RSD为0.1%(n= 8),表明该方法稳定性良好。
2.3.6 检出限 取掺伪比例为5%的供试品溶液,逐级稀释至特有峰信噪比为8.2;相当于以Gh1作为伪品,掺伪比例为0.5%时,为检出限。
2.3.7 定量限 取掺伪比例为5%的供试品溶液,逐级稀释至特有峰信噪比为9.3;相当于以Gh1作为伪品,掺伪比例为0.75%时,为定量限。
2.3.8 耐用性 比较了Agilent 5TC C18(4.6 mm ×250 mm,5 μm)、Waters X-Bridge TM C18(4.6 mm ×250 mm,5 μm)、日立LaChrom C18(4.6 mm × 250 mm,5 μm)等不同品牌色谱柱;不同柱温28、30、32℃;及Agilent 1260型、Waters ARC型不同高效液相色谱仪时该方法的耐用性。结果在皂角刺正品中均未检出特有峰,在野皂角刺中均检出特有峰,与阿魏酸的RRT约为1.43,表明该方法耐用性良好。
2.4 结果判定
选取3批皂角刺,5批野皂角刺,按重量百分比(W/W)掺入伪品野皂角刺3%的比例,按照表3所示编号,同“2.2.4”配制供试品溶液,并加入阿魏酸内标溶液,使其终浓度约为1 μg/mL,每组掺伪样品平行2份,共计得到20份掺伪比例3%的皂角刺样品,按照拟定HPLC方法测定。将阿魏酸折算成浓度为1 μg/mL时的峰面积,计算特有峰与阿魏酸峰面积比值,公式如下:
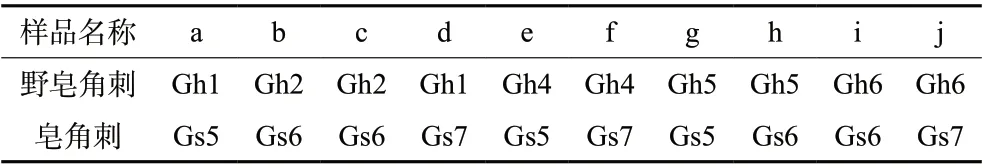
表3 掺伪3%比例的样品组成与编号Tab. 3 Sample composition and number of 3% adulteration ratio

其中,各参数如下:P比为特有峰/阿魏酸峰面积之比;A1为特有峰峰面积;A2为阿魏酸峰面积;C2为阿魏酸内标溶液浓度(单位: μg/mL);1代表阿魏酸浓度为1 μg/mL。
结果显示,当掺伪比例为3%时,20份掺伪样品特有峰/阿魏酸峰面积之比为0.18~0.80,色谱图见图4。在饮片实际加工过程中,可能达到的最低掺伪比例不会低于3%,同时参考《中国药典》2020年版(通则0212)中关于药材杂质不得过3%的规定,将皂角刺中掺入野皂角刺3%及以上时,认定为野皂角刺阳性检出。考虑到收集的野皂角刺样品来源有限,并避免某些样品在贮存运输中的意外污染而产生误判,将特有峰/阿魏酸峰面积比值高限值由0.80扩大至1.0。
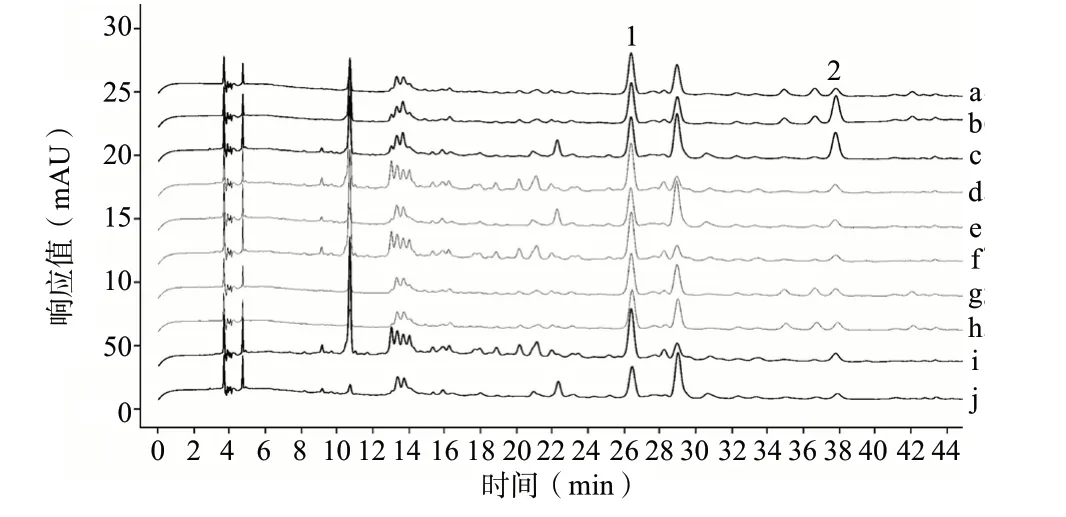
图4 掺伪3%比例样品色谱图Fig.4 The chromatogram of adulteration 3 % ratio samples
结果判定:供试品溶液色谱中,若出现与阿魏酸色谱峰相对保留时间为1.43的色谱峰,采用二极管阵列检测器比较阳性对照溶液中特有峰与该色谱峰在190~400 nm波长范围内紫外-可见光吸收光谱,二者应不相同;若吸收光谱相同,且该色谱峰与阿魏酸峰面积之比大于1.0,则视为野皂角刺阳性检出。
2.5 样品的测定
对收集到的26批皂角刺样品,经甘肃省药品检验研究院宋平顺主任药师鉴定,其中掺伪样品5批,掺伪检出率为19.2%。按照拟定HPLC法进行测定,结果7批样品检出野皂角刺;19批中未检出野皂角刺,掺伪检出率为26.9%,结果见表4。
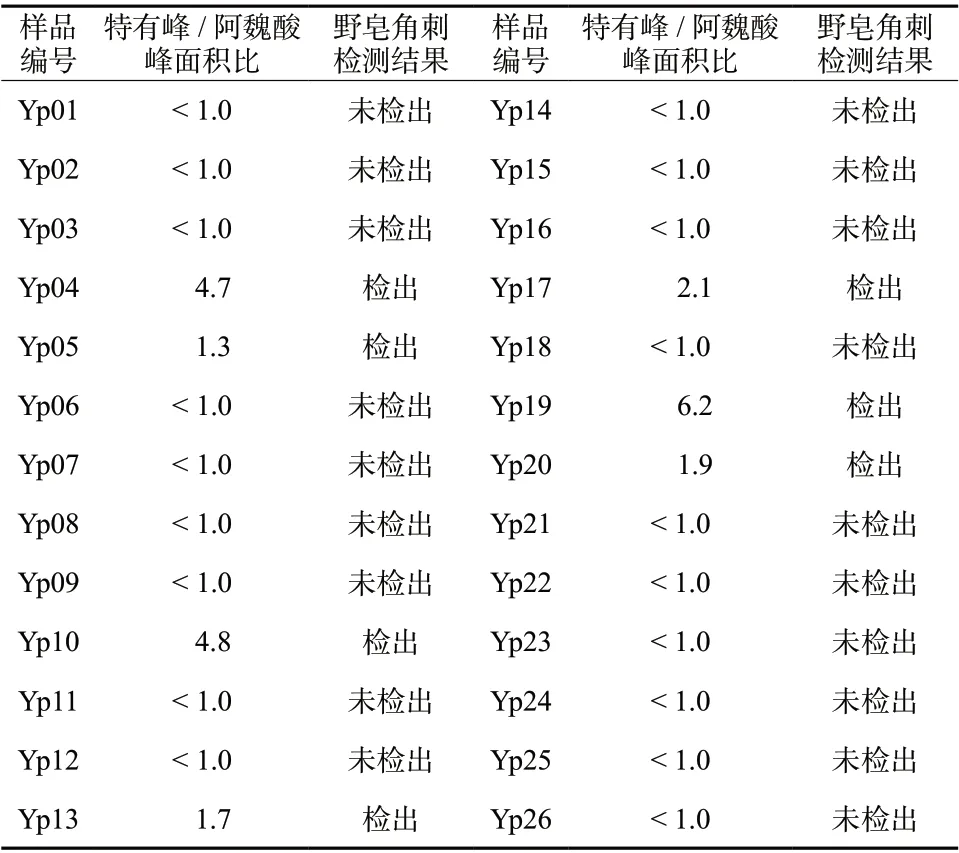
表4 皂角刺样品中野皂角刺检测结果Tab. 4 The determination results of Gleditsia heterophylla Bunge. in Gleditsia sinensisLam. samples
3 讨论
3.1 测定方法的建立
文献中皂角刺提取方法多是关于多组分测定及指纹图谱研究[13-15],如回流提取法、冷浸法及萃取法等。而本研究的目的是找出伪品野皂角刺中的特有成分。研究中对甲醇超声法及回流提取法提取样品进行了比较分析,并考察了提取溶剂、超声时间、提取时间等因素的影响。色谱条件参考了文献[13]:乙腈-0.1%甲酸为流动相,C18为色谱柱。方法建立过程中重点对流动相色谱洗脱程序进行了优化,以确定系统适应性达到要求,并排除假阳性干扰。预试验结果显示皂角刺与野皂角刺HPLC图谱有较大差异,野皂角刺图谱中可见一特有峰,其在190~400 nm范围内最大吸收波长为328 nm。研究中在保证特有峰响应灵敏度的前提下,正品皂角刺中的可干扰杂质峰去除为原则,最终选定的测定波长为305 nm。
3.2 内标法的确定
在本方法中,对皂角刺样品中是否含有特有峰进行准确定性分析是该方法是否可行的关键。关于皂角刺中化学成分及药理作用的相关文献多有报道[5,16-17],而对于伪品野皂角刺中化学成分无可参考资料。本研究显示特有峰含量的高低直接与皂角刺中野皂角刺的掺伪比例呈正相关,而该组份仅存在于伪品野皂角刺中,可见该特有峰并无皂角刺相关的药理作用,对其进行结构确证意义一般。研究中所拟HPLC方法用于鉴别皂角刺中掺入野皂角刺仍然可行。
因此,笔者利用加入的一种内标物质,对未知组分特有峰采用色谱RRT及紫外光谱进行联合定性。在筛选了多种化学组分后,最终确定以阿魏酸对照品作为内标物,特有峰相对于阿魏酸的RRT为1.43,介于0.5~1.5之间。经稳定性试验显示,阿魏酸在加入至皂角刺供试品溶液后,在24 h内的峰面积RSD为0.6% (n= 8),保留时间RSD为0.2% (n= 8)。表明以阿魏酸对照品作为内标物进行野皂角刺中特有峰的检测,稳定性好,简便易得,方法可行。
3.3 阳性检出限值的判断
研究中以掺伪3%野皂角刺的测定结果作为阳性检出限值,样品测定结果阳性检出率较之经验鉴别高出了7.7%,方法灵敏度较高。鉴于此次仅收集到6批伪品野皂角刺,其来源、产地并未达到全国范围野皂角刺生长区域全覆盖,对于特有峰/阿魏酸峰面积比测定确定的限度值存在一定的局限性。因此,对于阳性检出限值还需要进一步采集更多的伪品野皂角刺进行确认,以免出现假阳性而产生误判。
4 结论
本研究建立的HPLC-DAD内标法为快速鉴别皂角刺中掺入野皂角刺的情况提供了实验基础,可作为皂角刺质量评价的补充检验方法,为市场上皂角刺药材及饮片的质量监督提供依据。采用加入的一种内标物对伪品中特有峰进行定性,为一个新方法新思路,省去了结构确证等耗时耗资的过程,也可为市场其它药材掺伪掺假分析提供参考。
