Paramyrothecium roridum中单端孢霉烯毒素生物合成基因启动子的克隆和功能鉴定
2021-09-14岑由飞朱牧孜叶伟李赛妮钟国华章卫民
岑由飞 朱牧孜 叶伟 李赛妮 钟国华 章卫民
(1. 华南农业大学 农业农村部华南作物有害生物综合防治重点实验室 天然农药与化学生物学教育部重点实验室,广州510642;2. 广东省微生物研究所 广东省科学院 华南应用微生物国家重点实验室 广东省菌种保藏与应用重点实验室 广东省微生物应用新技术公共实验室,广州 510070)
单端孢霉烯族毒素主要是由镰刀菌、单端孢霉、木霉和其他霉菌产生的生物活性和化学结构相似的有毒代谢产物,可污染小麦、大麦、玉米等禾谷类作物,给农业带来巨大危害[1-2],同时,研究还发现单端孢霉烯毒素具有抗肿瘤、抗真菌、抗疟疾和植物毒活性[3-6],在药物开发方面具有良好的应用前景。据报道,tri3、tri4、tri5、tri6、tri11、tri12是单端孢霉烯生物合成的关键基因[7-9]。启动子是能够准确与RNA聚合酶识别、结合和开始转录的一段DNA序列,是基因转录最重要的功能组件。基因表达水平受多个因素的影响,其中启动子的强弱是一个重要的因素,基因表达水平高低与启动子启动活性强弱直接相关[10]。因此,启动子活性的强弱决定了基因的转录效率,从而决定细胞中mRNA的总体丰度和目标蛋白的含量[11-12]。近年来,人们不断从丝状真菌中发掘出具有很好生物活性的新型次级代谢产物以及生物酶,但是这些次级代谢产物以及生物酶的产量在原始菌株中往往很低,为了提高产量,不少丝状真菌的强启动子相继被发掘,如里氏木霉(Trichoderma reesei)的cbh1、pdcl、tef1启动子,橡胶树白粉菌(Oidium heveae)的WY193启动子,疣孢青霉(Penicillium verruculosum)的xylA启动子,木霉(Trichoderma sp.)的pki1启动子,构巢曲霉(Aspergillus nidulans)的glaA、trpC启动子等[13-14]。 启动子的启动效率与启动子中的功能组件密切相关,常见的启动子功能组件有TATA box,CAAT box和GC box。TATA box能指导起始前复合物的形成,决定转录起始位点,并调控上游激活蛋白的行为,CAAT box可以提高启动子的启动效率,GC box是一个转录调节区,有激活转录的功能[15]。据报道,tri5和tri12分别编码倍半萜环化酶和MFS协同超家族转运蛋白,是单端孢霉烯类化合物生物合成和外排过程中的关键酶[7-8]。
本课题组前期通过荧光定量PCR和琼脂糖凝胶电泳分析单端孢霉烯生物合成相关基因tri3、tri4、tri5、tri6、tri11、tri12的表达量,结果显示tri5的表达量最高,tri12的表达量次之,推测tri5和tri12的启动子可能具有较强的启动能力[16]。本研究采用染色体步移技术对P. roridum A553中单端孢霉烯毒素的生物合成关键基因tri5和tri12启动子进行克隆,采用氨苄青霉素抗性和潮霉素抗性基因表达载体进行功能验证;同时,为寻找启动子的核心区域,进一步利用荧光素酶载体对启动子核心区域进行定量分析,并利用软件对启动子的功能组件进行预测和分析,为后期通过转录调控和异源表达以获得更多高活性的单端孢霉烯毒素奠定分子生物学基础。
1 材料与方法
1.1 材料
1.1.1 仪器和试剂 通用型DNA纯化回收试剂盒、质粒小提试剂盒(天根生化试剂公司,北京),pEASY-T1 Cloning Kit、DNA Marker 2k(全式金生物技术有限公司,北京),Genome walking kit、PrimeSTAR MAX Premix(TaKaRa,大连),2× taq master mix(Microanalysis生物科技公司,北京),ClonExpress II One Step Cloning Kit C112(诺唯赞生物科技有限公司,南京)。
1.1.2 菌株和载体 植物内生真菌Paramyrothecium roridum A553分离自药用植物广藿香[5],pET30a载体、YEp352载体、pGL3-basic载体(淼灵生物科技有限公司,武汉),DH5α感受态细胞(全式金生物技术有限公司,北京),酿酒酵母BJ5464,保藏于广东省微生物研究所。
1.2 方法
1.2.1 tri5和tri12启动子的克隆和序列分析 基于前期的转录组测序结果,根据Genome walking kit试剂盒说明书,设计tri5和tri12启动子序列的特异性反向引物(表1)。以菌株A553基因组DNA为模板,利用通用正向引物和特异性反向引物AP3进行3次巢式PCR反应,并将巢式PCR产物进行琼脂糖凝胶电泳鉴定。将最后一步的PCR产物纯化回收,回收产物利用pEASY-T1试剂盒进行TA克隆,转化DH5α大肠杆菌感受态细胞,涂布于氨苄青霉素抗性平板筛选阳性克隆,利用通用引物T7和T7-ter进行菌落PCR筛选阳性克隆并提质粒测序加以验证,获得目的基因tri5和tri12上游相关启动子片段。将巢式PCR获得的相关启动子序列在启动子预测网站(https://www.fruitfly.org/seq_tools/promoter.html)进行分析获得启动子序列,并预测启动子的核心区域。

表1 目的基因tri5和tri12特异性反向引物序列Table 1 Specific reverse primer sequences of the target genes tri5 and tri12
1.2.2 启动子-pET30a-Amp、启动子-YEp352-HYRB-CYC1和启动子-pGL3-basic载体构建 为了寻找tri5和tri12启动子的核心区域,以1.2.1克隆得到的启动子片段为模板,设计同样的反向同源臂引物和不同的正向同源臂引物,通过PCR得到tri5和tri12启动子不同长度的片段,分别记为5-f0、5-f1、5-f2和12-f0、12-f1、12-f2。
启动子-pET30a-Amp载体构建:设计针对pET30a载体的同源臂引物,利用PrimeSTAR MAX Premix酶,以pEASY-T1载体为模板进行PCR得到Amp片段,同时扩增pET30a-ACP载体作为backbone,纯化回收PCR产物,利用ClonExpress II One Step Cloning Kit C112(Vazyme)将Amp片段和pET30a-ACP同源重组连结,转化DH5α感受态细胞后涂布于卡那霉素抗性平板筛选阳性克隆,通过菌落PCR和提质粒测序验证,获得带有氨苄青霉素抗性的pET30a-Amp空载。设计针对pET30a-Amp空载的同源臂引物,以1.2.1克隆得到的启动子为模板扩增得到6个片段,同时利用限制性内切酶Nde I和Xho I对pET30a-Amp空载进行双酶切,通过琼脂糖凝胶电泳鉴定,将PCR产物和酶切产物进行切胶回收并测定其浓度。利用同源重组试剂盒将6个启动子片段分别与pET30a-Amp空载体进行同源重组连结,转化DH5α感受态细胞后涂布于卡那霉素抗性平板筛选阳性克隆,通过菌落PCR和提质粒测序验证。
启动子-YEp352-HYRB-CYC1载体构建:设计针对pFC332-HYRB载体的同源臂引物,以pFC332-HYRB载体为模板扩增HYRB基因,同时扩增YEp352-TEF1-Amp载体作为backbone,纯化回收PCR产物,将其进行同源重组连结,得到YEp352-TEF1-HYRB-CYC1载体。利用限制性内切酶Xba I和Sal I对YEp352-TEF1-HYRB-CYC1载体进行双酶切除去TEF1启动子,同时设计针对ddYEp352-HYRB-CYC1载体的同源臂引物,以1.2.1克隆得到的启动子为模板扩增得到5-f0和12-f1启动子片段,通过琼脂糖凝胶电泳鉴定,将这两个片段与ddYEp352-HYRB-CYC1载体进行切胶回收,利用同源重组试剂盒分别将这两个片段与ddYEp352-HYRB-CYC1载体重组连结,转化DH5α感受态细胞涂布于氨苄青霉素抗性平板筛选阳性克隆,通过菌落PCR和提质粒测序验证。将测序结果正确的重组载体转化酵母感受态细胞BJ5464,再通过菌落PCR验证。
启动子-pGL3-basic载体构建:设计针对pGL3-basic载体的同源臂引物,以1.2.1克隆得到的启动子片段为模板扩增得到5-f0和12-f2启动子片段,同时利用限制性内切酶NdeI和HindIII对pgpd-pGL3-basic载体(pgpd是真菌中已鉴定的强启动子[17])进行酶切,通过琼脂糖凝胶电泳鉴定,将PCR产物和酶切产物进行切胶回收,获得启动子的5-f0、12-f2启动子片段和ddpGL3-basic。利用重组试剂盒将这2个片段分别与ddpGL3-basic同源重组后,转化DH5α感受态细胞涂布于氨苄青霉素抗性平板筛选阳性克隆,通过菌落PCR和提质粒测序验证。
1.2.3 氨苄青霉素抗性基因表达验证 将1.2.2得到的DH5α-pET30a-5-f0/5-f1/5-f2/12-f0/12-f1/12-f2-Amp 转化子以及DH5α-pET30a-ACP空载(阴性对照)接种于含卡那霉素的LB液体培养基,将DH5α-YEp352-12-f1-HYRB-CYC1(阳性对照)接种于含氨苄青霉素的LB液体培养基,37℃过夜培养,取过夜培养的菌液稀释至OD600值为 1.0左右,取出10 μL加入到990 μL无菌水中稀释100倍,记为10-2,然后从10-2取出10 μL加入到90 μL无菌水中稀释10倍,记为10-3,再从10-3取出10 μL加入到90 μL无菌水中稀释10倍,记为10-4,从不同浓度菌液中各取5 μL点在氨苄青霉素浓度为0、50、75 μg/mL的LB平板上,37℃培养10-12 h,观察培养皿中各菌落的生长情况并拍照。根据点板结果,将阳性菌株与长出的菌落数与阳性菌株无差异的菌株分别接种于对应的抗性培养基中培养12 h,利用含有氨苄青霉素抗性的LB培养基作为稀释液,按上述方法进行稀释,37℃,180 r/min培养。将不同稀释浓度的菌液加至96孔板中,每孔200 μL,每个稀释菌液设置五个重复,用Thermo多功能酶标仪测定菌株的OD600值。每隔1 h测定1次,共10次,实验重复3次。利用GraphPad Prism 8.0.1对相同浓度稀释菌液的不同菌株的生长OD值进行显著性分析。
由于上述点板结果不能比较3个tri5启动子片段的转录活性,故重新培养DH5α-pET30a-ACP空载(阴性对照)、DH5α-YEp352-12-f1-HYRB-CYC1(阳性对照)和DH5α-pET30a-5-f0/5-f1/5-f2-Amp系列转化子,再按上述方法稀释,各取5 μL点在氨苄青霉素浓度为0、10、25 μg/mL的LB平板上,37℃培养10-12 h,观察培养皿中各菌落的生长情况并拍照。
1.2.4 潮霉素抗性基因表达验证 将1.2.2得到的BJ5464-YEp352-HYRB-CYC1空载(阴性对照)、BJ5464-YEp352-TEF1-HYRB-CYC1(阳 性 对 照)、BJ5464-YEp352-5-f0-HYRB-CYC1、BJ5464-YEp352-12-f1-HYRB-CYC1接种于SD-Ura液体培养基中,30℃培养24 h,传代一次再培养24 h。将菌液按1.2.3的方法进行稀释,各取5 μL点在0、75、100 μg/mL潮霉素抗性的YPD平板上,30℃培养48 h,观察培养皿中各菌落的生长情况并拍照。根据点板结果,将阳性菌株与长出的菌落数与阳性菌株无差异的菌株分别接种于对应的SD-Ura培养基中培养24 h,利用含有潮霉素抗性的SD-Ura培养基作为稀释液,按1.2.3的方法进行稀释,30℃,180 r/min培养。将不同稀释浓度的菌液加至96孔板中,每孔200 μL,每个稀释菌液设置5个重复,用Thermo多功能酶标仪下测定生长OD600值。每隔6 h测定1次,共8次,实验重复3次。利用GraphPad Prism 8.0.1对相同浓度稀释菌液的不同菌株的生长OD值进行显著性分析。
1.2.5 启动子启动荧光素酶基因的表达 将构建成功的含有pGL3-5-f0-basic和pGL3-12-f1-basic质粒载体和pGL3-pgpd-basic载体的菌株分别接种于含有1.0 mL LB液体培养基的EP管中,37℃,200 r/min培养10-12 h,按1∶100传代到10 mL LB培养基,继续培养3-5 h,测定OD600值。当OD600值为0.8-1.0时,离心收集菌液,并用PBS缓冲液洗涤2-3次,最后用500 μL的PBS重悬,利用手持细胞超声仪破碎(振幅 30%,工作3 s/停顿10 s)20 min,12 000 r/min离心2 min,分离上清和沉淀。分别取50 μL上清和50 μL萤火虫荧光素酶检测试剂于96孔板中,快速混匀后立即使用Tecan Spark多功能酶标仪检测各反应的RU值,反应时间为10 s。
1.2.6 tri5和tri12启动子核心区域分析 利用在线PlantCARE数据库预测tri5和tri12启动子转录相关的功能组件。结合点板结果,预测启动子的核心 区域。
2 结果
2.1 tri5启动子和tri12启动子的克隆分析
通过3次巢式PCR反应对P. roridum A553基因组扩增,得到tri5和tri12对应的扩增产物G1、G2和G3,tri5和tri12最后一次巢式PCR扩增产物G3的目的条带大小分别约为2.5 kb(图1-A)和2.8 kb(图1-B)。纯化回收最后一次巢式PCR产物,利用pEASY-T1试剂盒进行TA克隆,转化至DH5α感受态细胞,涂布于氨苄青霉素抗性平板筛选出阳性克隆,菌落PCR验证阳性克隆并测序,获得tri5和tri12的相关启动子片段,利用启动子预测网站(https://www.fruitfly.org/seq_tools/promoter.html)预测得到的启动子序列,tri5和tri12启动子大小分别为1 458 bp和1 356 bp。将tri5和tri12启动子序列在网站上进行核心区域分析,分别在网站(https://www.cbs.dtu.dk/services/Promoter/)和(https://www.fruitfly.org/seq_tools/promoter.html)预测tri5和tri12启动子的核心区域,分值相对较高(表2)。

表2 目的基因tri5、tri12启动子核心序列Table 2 Core sequences of tri5 and tri12 promoters
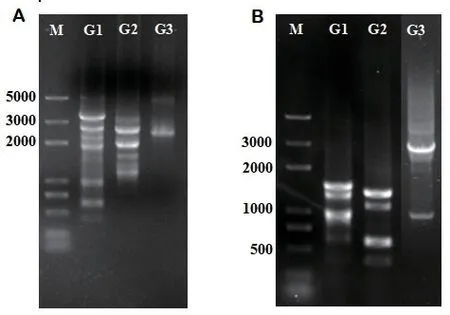
图1 tri5、tri12染色体步移扩增得到的目的片段Fig.1 Target fragments of tri5 and tri12 obtained by chromosome walking
2.2 启动子-pET30a-Amp质粒的构建及氨苄青霉素抗性的验证
设计同源臂引物将tri5和tri12启动子分别分成3个片段(图2),与pET30a-ACP空载体同源重组得到重组载体,转化大肠杆菌感受态细胞,涂布于卡那霉素抗性平板筛选阳性克隆,通过菌落PCR验证并测序成功(图3-A),得到启动子-pET30a-Amp载体(图3-B),即得到对应的DH5α-pET30a-5-f0/5-f1/5-f2/12-f0/12-f1/12-f2-Amp菌株。将这6个菌株及DH5α-pET30a-ACP空载(阴性对照)与DH5α-YEp352-12-f1-HYRB-CYC1(阳性对照)接种于对应的培养基,37℃培养12 h,将菌液稀释后分别点在氨苄青霉素抗性浓度为0、50、100 μg/mL的LB平板上,37℃培养12 h。发现在100 μg/mL氨苄青霉素抗性板上只有DH5α-pET30a-12-f1-Amp和DH5α-YEp352-12-f1-HYRB-CYC1长出菌落(图4-A),通过测定生长曲线和显著性分析发现,相同稀释浓度的DH5α-pET30a-12-f1-Amp和DH5α-YEp352-12-f1-HYRB-CYC1的生长OD值没有显著性差异(P>0.05,图4-C),说明tri12启动子片段12-f1和阳性对照启动子的启动效率均比tri5启动子片段5-f0强,而tri12启动子片段12-f1的启动效率与阳性对照启动子相当。

图2 构建具有不同长度片段的tri5和tri12基因启动子Fig.2 tri5 and tri12 promoters with different lengths of fragments
为了确定3个tri5启动子片段的启动效率,重新培养含tri5启动子片段的菌株以及DH5α-pET30a-ACP空载(阴性对照)和DH5α-YEp352-12-f1-HYRB-CYC1(阳性),将菌液稀释后分别点在氨苄青霉素抗性浓度为0、10、25 μg/mL的LB平板上,37℃培养12 h。发现在25 μg/mL的平板上只有DH5α-pET30a-ACP空载(阳性对照)和DH5αpET30a-5-f0-Amp长出菌落(图4-B),且菌落数较多,说明tri5的启动子片段中,5-f0的启动效率最强。

图4 不同浓度的氨苄青霉素抗性平板筛选含不同启动子片段的大肠杆菌Fig.4 Screening of E. coli containing different promoter fragments on resistance plates with different concentrations of ampicillin
2.3 启动子-YEp352-HYRB-CYC1质粒的构建及潮霉素抗性的验证
设计针对YEp352载体的同源臂引物,将tri5和tri12启动子分别分成3个片段,与双酶切的YEp352-HYRB-CYC1载体进行同源重组连结,转化大肠杆菌感受态细胞,涂布于氨苄青霉素抗性平板筛选阳性克隆,通过菌落PCR验证并测序成功(图3-C),得 到YEp352-5-f0/5-f1/5-f2/12-f0/12-f1/12-f2-HYRB-CYC1载 体(图3-D)。将YEp352-TEF1-HYRB-CYC1(阳 性 对 照)、YEp352-No pro-HYRB-CYC1(阴 性 对 照)、YEp352-12-f1-HYRBCYC1和YEp352-5-f0-HYRB-CYC1质粒转化酵母感受态细胞BJ5464。通过菌落PCR和测序验证,结果表明这4个载体都成功转入酵因细胞中。将成功转入YEp352系列载体的酵母菌株分别接种至SDUra液体培养基中,30℃培养2 d,将菌液稀释后分别点在潮霉素浓度为0、25、50、75 μg/mL的YPD平板上,37℃培养2 d,发现在75 μg/mL的YPD上BJ5464-YEp352-5-f0-HYRB-CYC1的菌落数极少,而BJ5464-YEp352-12-f1-HYRB-CYC1和BJ5464-YEp352-TEF1-HYRB-CYC1的菌落数较多(图5-A),通过测定生长曲线和显著性分析发现,相同稀释浓度的BJ5464-YEp352-12-f1-HYRB-CYC1和BJ5464-YEp352-TEF1-HYRB-CYC1的生长OD值均没有显著性差异(P>0.05,图5-B),说明tri12启动子片段12-f1和阳性对照启动子的启动效率均比tri5启动子片段5-f0强,而tri12启动子片段12-f1的启动效率与阳性对照启动子相当。
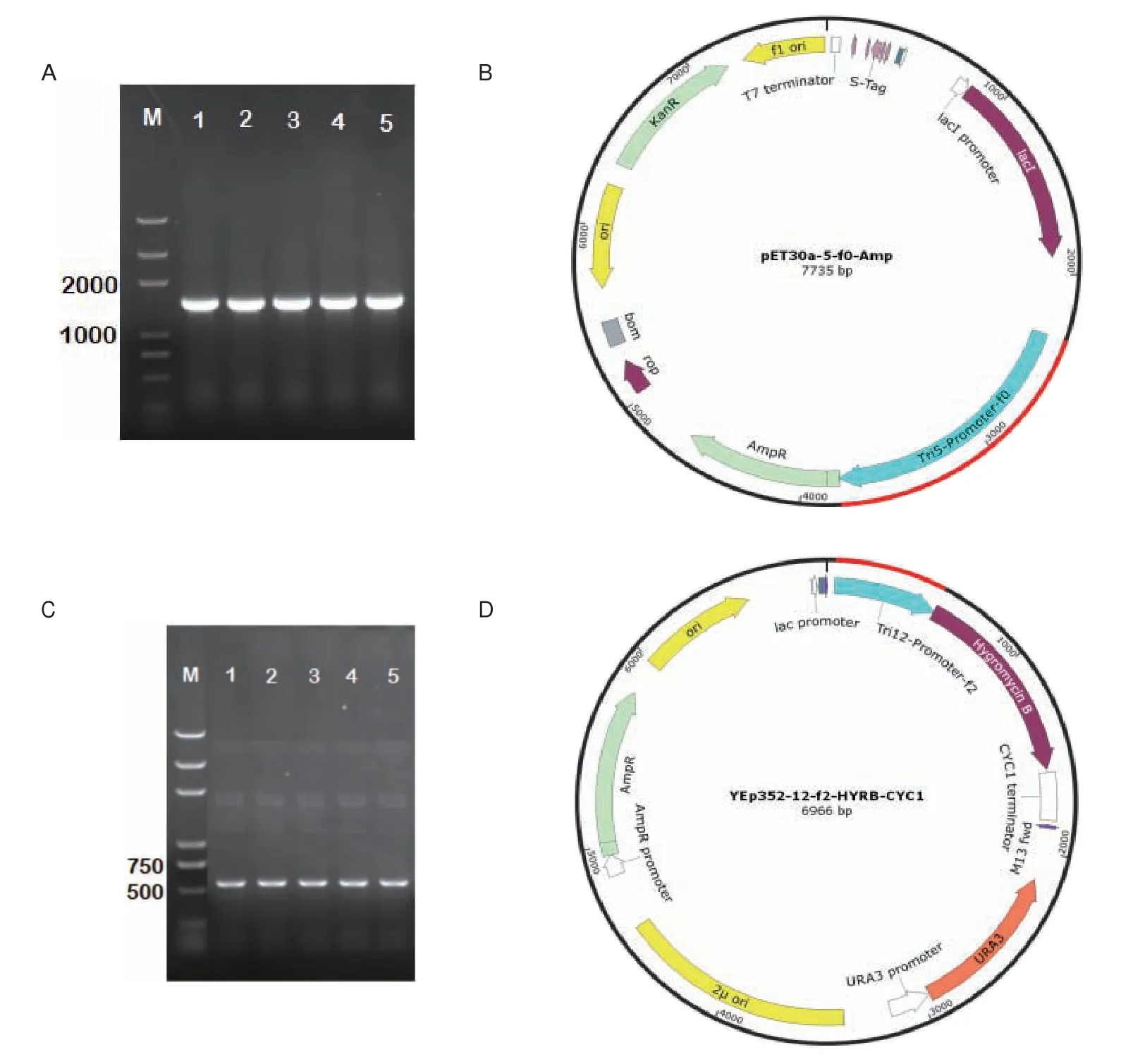
图3 DH5α菌落PCR验证及启动子载体Fig.3 Colony PCR verification of DH5α and promoter vector maps

图5 不同浓度的潮霉素抗性平板筛选含不同启动子片段的酿酒酵母Fig.5 Screening of S. cerevisiae containing different promoter fragments on resistant plates with different concentrations of hygromycin
2.4 荧光素酶活性检测
利用同源重组的方法成功构建了分别含有5-f0、12-f0启动子片段的pGL3-basic重组表达载体,转化DH5α大肠杆菌感受态细胞,并通过菌落PCR和测序验证。利用荧光素酶检测试剂对成功导入pGL3-basic重组载体的重组菌进行检测。结果(图6)显示含有pgpd启动子的菌株其荧光素酶活性均高于含有5-f0和12-f0启动子片段的重组菌,含有5-f0启动子片段的重组菌的荧光素酶活性高于含有12-f1启动子片段的重组菌,说明5-f0启动子片段对荧光素酶基因的启动效率高于12-f1启动子片段。这与点板的结果相反,说明同一个启动子在启动不同基因时的启动效率不同。
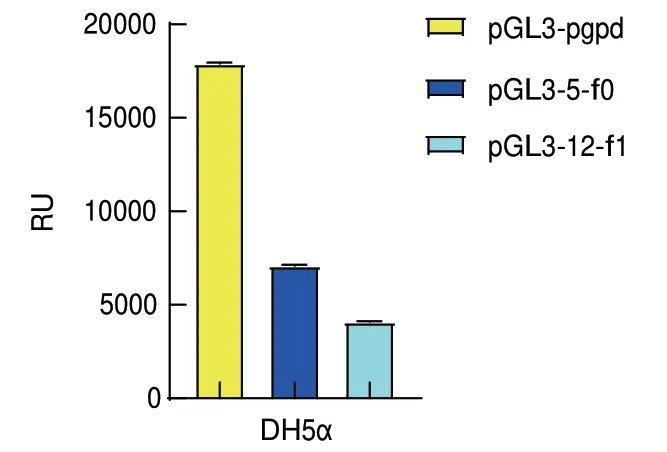
图6 pGL3-5-f0-basic、pGL3-12-f1-basic和pGL3-pgpdbasic载体在DH5α中的荧光素酶活性检测Fig.6 Luciferase activity detection of pGL3-5-f0-basic,pGL3-12-f1-basic and pGL3-pgpd-basic vectors in DH5α
2.5 tri5和tri12启动子活性区域分析
利用PlantCARE数据库预测启动子的功能组件,结合启动子序列特点进行分析发现,在tri5启动子的-43处有一个TATA box,使转录精确起始;在启动子的-66处有一个CAAT box,控制转录起始频率。在tri12启动子的转录起始位点的上游30-50 bp处并没有TATA box,因此认为该启动子是一个TATAless启动子;但在启动子的-54处有一个CAAT box,控制转录起始频率;在CAAT-box前面与其相连有一个GC box,起到激活转录的作用(图7)。结合点板的结果分析,片段5-f0启动活性强于5-f1,5-f2,说明tri5启动子的ATG上游941-1 458 bp之间存在正向调控的功能组件,可增强启动活性。片段12-f1的启动活性显著高于12-f0和12-f2,表明在tri12启动子的ATG上游999-1 356 bp之间和1-516 bp之间存在转录抑制因子,影响启动活性;而ATG上游516-999 bp之间存在正向调控的功能组件,可增强启动活性,即说明片段12-f1就是tri12启动子的核心区域。
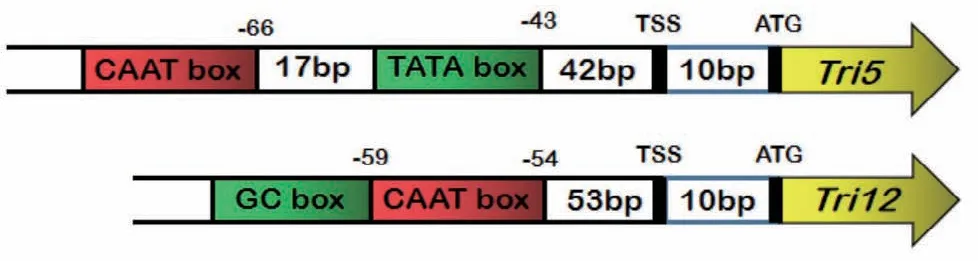
图7 tri5和tri12功能组件预测Fig.7 Functional component prediction of gene tri5 and tri12
3 讨论
丝状真菌中次级代谢产物的生物合成需要一些关键酶的调控,而转录效率高的启动子可以启动关键酶的高效表达进而促进次级代谢产物的高效生物合成[17]。丝状真菌中的许多强启动子已经用于内源或外源蛋白的表达,例如构巢曲霉甘油醛3磷酸脱氢酶启动子(PgpdA)用于异源表达治疗性蛋白质-人干扰素β(HuIFNβ)[18],里氏木霉CBH1启动子用于异源表达纤维二糖水解酶Cel7A[19]。单端孢霉烯毒素是一类抗肿瘤活性显著的倍半萜类化合物,其生物合成基因簇已被许多文献报道,其中tri5、tri12是单端孢霉烯合成家族tri簇中不可或缺的关键基因。tri5编码的倍半萜环化酶是单端孢霉烯生物合成中的关键酶,可通过转录调控提高单端孢霉烯类化合物的产量。tri12不直接参与单端孢霉烯的生物合成,但其编码的MFS协同超家族转运蛋白在毒素外排的过程中起着关键性作用,防止毒素在细胞内过量积累而导致细胞死亡,毒素只有源源不断地外排,才能源源不断地在细胞内合成。尽管已报道了不少单端孢霉烯生物合成基因,但这些基因的启动子却很少被研究。
本研究对露湿漆斑菌中tri簇关键基因tri5和tri12启动子进行了克隆和功能鉴定,发现两者都具有良好的转录活性,提示其启动子在单端孢霉烯生物合成中发挥着重要作用。氨苄青霉素抗性基因和潮霉素抗性基因分别是原核和真核表达系统中最常见的抗性筛选基因,因此,本课题组利用这两种抗性基因对tri5和tri12启动子的核心区域进行鉴定[20],在后续的研究中还可将启动子整合至酿酒酵母染色体上进一步验证其活性。为了寻找启动子核心区,本研究将tri5和tri2启动子进行分段克隆,发现片段12-f1的转录活性比其他片段的转录活性强,因此可通过改造启动子而增强其活性。同时,通过对tri5和tri2启动子的克隆以及在酵母细胞中的表达,发现这两种启动子也能在酵母细胞中调控潮霉素抗性基因的转录和表达,说明它们在原核生物和真核生物中均具有启动活性,提示这两种启动子可用于不同宿主异源表达单端孢霉烯的高效生物合成。本研究为后期通过对P. roridum A553中单端孢霉烯毒素生物合成基因进行转录调控和异源表达奠定分子生物学基础。
4 结论
本研究对植物内生真菌Paramrothecium roridum A553中tri5和tri2两个基因的启动子序列进行克隆,采用原核和真核表达系统以及荧光素酶表达系统对tri5和tri2启动子的核心区域进行分析,发现tri5和tri2启动子具有较强的启动基因表达的能力且能够在酵母细胞中调控潮霉素基因的异源表达。最后利用软件分析预测了启动子中的关键功能组件。
