对乙酰氨基酚致药物性肝损伤的分子机制
2021-09-13王帆朱哿瑞刘成海陶艳艳
王帆 朱哿瑞 刘成海 陶艳艳
药物性肝损伤(drug-induced liver injury,DILI)是目前最常见以及最严重的药物不良反应之一,重者可致肝衰竭甚至是死亡;据世界卫生组织统计,每100 000病人至少有10~15例DILI[1]。作为临床常用药物,对乙酰氨基酚(Acetaminophen,APAP)仍是许多国家引起DILI和急性肝衰竭的常见原因[2]。通常APAP诱导的氧化应激和线粒体功能障碍在DILI的发病机制中起着核心作用[3],近年来,APAP肝毒性有了更深入的研究,涉及APAP和/或其代谢产物导致丝裂原活化蛋白激酶(mitogen activated protein kinase,MAPK)激活、线粒体膜通透性转变(mitochondrial membrane permeability transition,MPT)、Keap1-Nrf2-ARE激活等多种信号分子传导途径,本文旨在综述APAP肝损伤的分子机制,为其临床治疗拓展思路。
一、APAP在肝脏中的代谢
通常情况下,治疗剂量的APAP进入肝脏后,大部分(约90%)的APAP在II相代谢反应结合酶的作用下转化成无毒的化合物,并通过肾脏与尿液一同排出体外,参与此反应的结合酶主要是UDP-葡萄糖醛酸转移酶(UDP-glucuronyltransferase,UGTs)和磺酸转移酶(sulfotransferase,SULTs)。剩余的APAP(约有10%)被细胞色素P450酶(cytochrome P450,CYP450)代谢,从而形成反应性代谢产物N-乙酰基-P-苯醌亚胺(NAPQI),高反应性的NAPQI可快速与肝谷胱甘肽(Glutathione,GSH)结合并排入胆汁[4]。但当APAP达到毒性剂量时,过量的APAP将会导致II期代谢葡萄糖醛酸或硫酸化途径饱和,从而使大量的APAP进入到CYP途径中,继而增加了NAPQI的产生[5]。NAPQI的聚集会进一步导致还原型谷胱甘肽(GSH)的严重耗竭;在GSH耗尽后,过量的NAPQI将与替代目标(如蛋白质、DNA、不饱和脂质等)反应并产生一系列反应,如氧化应激、脂质过氧化等,最终导致肝细胞的死亡。同时,作为一种抗氧化酶,GSH的降低会加重肝细胞的氧化应激。氧化应激可形成大量的副产物ROS,通过改变DNA、脂质、蛋白质等大分子引起肝细胞的损伤。
总的来说,无论是NAPQI与蛋白质结合所形成的APAP蛋白质加合物(APAP Protein Adducts),抑或是GSH的耗竭都会引起氧化应激并最终发挥细胞毒作用。目前发现与氧化应激相关的分子通路主要有JNK/ASK-1[6]以及Nrf2[7]、TRPM2激活,且针对上述几种信号通路的抑制剂均可保护APAP肝损伤。另外,线粒体通透性改变(mitochondrial permeability transition,MPT)在APAP诱导的肝细胞损伤以及坏死中也有着不可忽略的作用,MPT已经被证明是APAP毒性机制中的重要一环[8]。
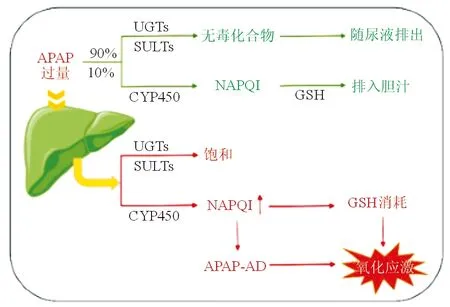
UGTs:葡萄糖醛酸转移酶;SULTs:磺酸转移酶;CYP450:细胞色素P450酶;GSH:谷胱甘肽;NAPQI:N-乙酰基-P-苯醌亚胺;APAP-AD:APAP蛋白质加合物图1 APAP在肝脏中的代谢
二、JNK信号通路激活
JNK(c-Jun N-terminal kinase,c-Jun氨基末端激酶)又称为应激活化蛋白激酶(stress-activated protein kinase,SAPK),属于丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)家族。JNK亚型——JNK1、JNK2、JNK3蛋白属于丝氨酸/苏氨酸蛋白激酶,主要存在于细胞质中[9],多种信号如紫外线照射、毒性剂等均可激活JNK信号通路,且JNK持续激活会促进细胞的损伤以及死亡。
毒性剂量APAP作用下,JNK信号通路的激活主要与氧化应激产生的ROS相关。其中,早期(1~2 h)阶段由糖原合酶激酶3β(glycogen synthase kinase-3β,GSK-3β)以及下游混合谱系激酶3(mixed-lineage kinase-3,MLK3)参与激活。有研究表明,在APAP诱导小鼠肝损伤的早期,使用反义寡核苷酸沉默GSK-3β,可以起到保护APAP肝损伤的作用[10]。晚期(2~4 h)阶段则受到肝脏中凋亡信号调节激酶1(apoptosis signal-regulating kinase-1,ASK-1)的调节,ASK-1为MAPK激酶激酶(MAPK kinase kinases,MAPKKKs)[11],其激活对JNK信号通路磷酸化起着主要作用。ASK-1与GSK-3β均可使MKK4/7磷酸化,从而激活JNK信号通路[12]。JNK信号通路激活后易位至线粒体并与Sab蛋白结合。研究表明,当Sab与JNK结合时,其位于线粒体外膜内侧的非受体型6 蛋白酪氨酸磷酸酶(SHP1)将会被磷酸化生成 p-SHP1并转移至线粒体内膜,进而通过内膜对接蛋白使 p-Src去磷酸化失活并形成具有破坏电子传递作用的Src,在线粒体呼吸链的作用下加剧氧化应激ROS的产生[13],从而进一步维持JNK活性,诱导线粒体膜通透性改变,从而导致线粒体肿胀坏死[12]。
目前最常用的JNK抑制剂为SP600125,当SP600125的浓度为20 μmol/L时可以有效保护APAP致原代肝细胞坏死[14]。Saito等[15]发现,SP600125可在促进肝脏GSH(还原型谷胱甘肽)恢复的同时降低肝脏线粒体GSSG(氧化型谷胱甘肽)水平,从而证明了SP600125对肝细胞的保护作用主要由于其对线粒体氧化应激抑制。来氟米特(Leflunomide,LEF)是临床常用的抗风湿类药物,同样对肝细胞具有保护APAP肝损伤作用。有研究表明,来氟米特可抑制JNK1、JNK2磷酸化,阻止线粒体通透性的改变及促细胞凋亡因子的释放,从而保护肝细胞损伤[16]。
三、Keap1-Nrf2-ARE信号激活
当氧化应激产生自由基时,机体中维持细胞氧化还原平衡的抗氧化防御系统会被激活。常见的抗氧化信号通路——Keap1-Nrf2-ARE通路,由Kelch样ECH相关蛋白1(Keap1)、核因子类红细胞2-相关因子2(Nrf2)以及抗氧化反应元件(ARE)组成[17]。Nrf2是一种可与抗氧化反应元件(antioxidant response element,ARE)结合的转录因子,属于CNC转录因子家族[18],其抗氧化作用的激活与Keap1相关。ARE作为一种顺式增强子序列,对下游靶基因表达的调控有重要作用[19]。氧化应激时,正常状态下的Keap1-Nrf2聚合物将会因为Keap1半胱氨酸残基的氧化而解离,致使Nrf2 进入细胞核内并与ARE结合,从而激活下游抗氧化酶(如血红素氧合酶-1、谷氨酸半胱氨酸连接酶)的转录[18]。Keap1-Nrf2-ARE对调节细胞氧化还原状态维持细胞稳态有着重要作用[20]。已有研究证明,柠檬苦素(Limonin)可通过激活Nrf2抗氧化途径来减轻APAP诱导的肝毒性[21]。另外,yan等[22]发现,天然产物穿心莲内酯(andrographolide)可通过激活Nrf2并增加其下游基因表达来减轻APAP引起的肝损伤。
四、TRPM2通道激活
瞬时受体电位阳离子通道亚家族M成员2(transient receptor potential cation channel subfamily M member 2,TRPM2)是位于细胞膜上的非选择性阳离子通道,具有二磷酸腺苷核糖(adenosine diphosphoribose,ADPR)水解酶活性。氧化应激时,活性氧激活聚ADPR聚合酶(PARP),进而诱导ADPR的产生和TRPM2通道激活,使Ca2+内流增加;采用SiRNA阻断TPMP2表达可保护APAP诱导肝细胞损伤;同样,与野生型小鼠相比,TRMP2-/-小鼠APAP肝毒性明显减轻,这表明TRMP2通道在APAP所致肝损伤中起着重要作用[23]。姜黄素(Curcumin)是一种新型的TRPM2通道抑制剂,可用于APAP肝损伤的治疗[24]。另一方面,活性氧激活CaMKII蛋白(Ca2+/calmodulin-dependent kinase II),最终导致BECN1蛋白的磷酸化[25]。BECN1蛋白包含Bcl-2同源性(BH3)结构域、卷曲螺旋结构域(CCD)和进化保守结构域(ECD),在自噬中起着至关重要的作用[26]。磷酸化的BECN1会与PIK3C3(Phosphoinositide-3-Kinase Class 3)分离抑制自噬,并与BCL2或BCL2L1(BCL2 Like 1)结合,从而导致BAX(与BCL2相关的X蛋白)与BCL2分离,诱导凋亡,使用CaMKII抑制剂KN-93治疗可减轻小鼠APAP肝损伤[27]。
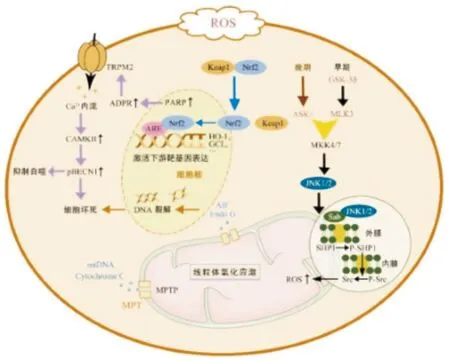
Keap1:Kelch样ECH相关蛋白1;Nrf2:核因子类红细胞2-相关因子2;ARE:抗氧化反应元件;HO-1:血红素氧合酶-1;GCL:谷氨酸半胱氨酸连接酶;Cytochrome C:细胞色素C;mtDNA:线粒体DNA;Endo G:线粒体蛋白内切核酸酶G;AIF:凋亡诱导因子;MPT:线粒体膜通透性转变;MPTP:线粒体膜通透性转换孔;GSK-3β:糖原合酶激酶3β;MLK3:下游混合谱系激酶3;ASK-1:凋亡信号调节激酶1;Sab蛋白:优先与Btk结合的SH3结构域结合蛋白;SHP1:非受体型6 蛋白酪氨酸磷酸酶;PARP:聚ADPR聚合酶;ADPR:二磷酸腺苷核糖;TRPM2:瞬时受体电位阳离子通道亚家族M成员2;CAMKⅡ:钙/钙调蛋白依赖性蛋白激酶Ⅱ图2 APAP毒性作用下主要分子机制
五、线粒体通透性改变
线粒体同样是APAP的作用靶点,当APAP与线粒体蛋白质结合时会导致线粒体电子传递链的紊乱以及线粒体氧化应激。氧化应激所产生的过氧化物、过亚硝酸盐等作用于线粒体时将会造成线粒体通透性的改变(mitochondrial permeability transition,MPT)。此时,一种非特异性孔——线粒体通透性过渡孔(MPTP)将会打开,使得线粒体对分子量小于1.5 kDa 的阴阳离子溶质的通透性突然增加[28]。
MPTP是一种由腺嘌呤核苷酸转位酶(ANT)、电压依赖性阴离子通道(VDAC)亲环蛋白D(cyclophilin D)构成的蛋白组合体[29]。钙离子积聚以及ROS均可导致MPTP的开放[30]。从而导致机制代谢物(最高不超过1 500 kDa)、线粒体蛋白内切核酸酶G(endonuclease G)、凋亡诱导因子(apoptosis inducing factor,AIF)以及线粒体DNA释放进入细胞质[31]。核酸内切酶G和AIF的释放可促进DNA裂解和细胞坏死,mtDNA可以触发TOLL样受体9(TLR9)诱导促炎性介质的表达和中性粒细胞的浸润以加重肝损伤[32]。
六、总结及展望
APAP造成的肝损伤有明显的剂量依赖性且作为机制较为复杂,除了目前较为明确的氧化应激,还可造成炎症反应[33]、内质网应激等,具体分子机制涉及丝裂原活化蛋白激酶(mitogen activated protein kinase,MAPK)激活、Keap1-Nrf2-ARE信号激活、线粒体膜通透性转变(mitochondrial membrane permeability transition,MPT)等等。
NAC(N-acetyl cysteine,N-乙酰-L-半胱氨酸)是含有巯基的抗氧化剂,体内代谢后去乙酰化生成半胱氨酸,并在肝细胞内进一步转化为谷氨酰半胱氨酸,然后在GSH合酶的作用下生成谷胱甘肽,从而达到解毒的作用。NAC虽然被广泛承认并且大量应用于临床APAP肝损伤治疗,但是其作用具有时间局限性,NAC对APAP导致肝毒性8 h内有较好的疗效,一旦超出时间范围,疗效将逐渐减弱[34]。同时,静脉注射NAC可导致静脉荨麻疹瘙痒等不良反应。因而,探究APAP肝毒性的分子机制仍有重要意义,并且有助于基于APAP的分子作用机制寻找潜在的治疗靶点,从而更好防治APAP药物性肝损伤的发生。
