遗传性凝血因子Ⅴ缺乏基因型和表型的关联研究
2021-09-09李可可陈朝霖
李可可,陈朝霖,冯 莹,肖 扬
广州医科大学附属第二医院血液内科,广州 510260
遗传性凝血因子Ⅴ缺乏(inherited factorⅤdeficiency,FⅤD)是一种罕见的常染色体隐性遗传性出血性疾病,发病率约为0.1/10万[1]。轻型FⅤD[FⅤ活性水平(FⅤ∶C)>10%],通常无临床症状;中型FⅤD(FⅤ∶C<10%)及重型FⅤD(FⅤ:C<1%),临床表现为程度不等的出血,如皮肤瘀斑、鼻衄、月经过多和术后出血增多等[2]。血浆FⅤ具有促凝和抗凝的双重作用,在止血和血栓的形成中均发挥重要作用[3]。研究[4-6]表明,血小板FⅤ也参与止血,并且与血浆FⅤ相比具有更强的促凝活性。FⅤD患者FⅤ∶C活性明显降低,但活性和临床出血程度并不一致,且临床出血表型也呈现出一定的异质性[4]。研究[7]发现正常血浆中FⅤ和组织因子途径抑制物(tissue factor pathway inhibitor,TFPI)以复合物的形式存在;FⅤD患者的TFPI水平降低,低TFPI水平可使FⅤD患者血浆中凝血酶生成的FⅤ最低需要量降至1%,可保护FⅤD患者免于严重出血。本研究通过对FⅤD患者进行临床表型诊断和基因检测,依照国际血栓与止血学会(International Society for Thrombosis and Hemostasis,ISTH)推荐的出血评分表对患者的出血风险进行评分,结合凝血酶生成试验(thrombin generation assay,TGA)综合评估患者的临床出血风险,以期探索FⅤD患者基因型和表型的关系。
1 对象与方法
1.1 研究对象
选取2020年1月—11月于广州医科大学附属第二医院血液内科门诊的5例家系FⅤD患者。纳入标准:①凝血酶原时间(prothrombin time,PT)、活化部分凝血活酶时间(activated partial thromboplastin,APTT)均延长,且其他凝血相关指标正常。②只有FⅤ活性水平(FⅤ∶C)低于50%。③有出血症状,如皮肤瘀斑、牙龈出血、鼻衄等。④无其他出血相关疾病。排除标准:①FⅤ和FⅧ活性均低于50%。②消耗性凝血障碍、肝病、FⅤ和FⅧ联合缺陷等疾病。③FⅤ抗体阳性。研究获医院伦理委员会批准(批准号:2020-YJS-ks-06)。
家系1:先证者,女,25岁。因咽痛就诊查凝血功能,发现PT、APTT延长,凝血因子FⅤ∶C为2.4%,其他因子均正常。皮肤易发瘀斑,无其他出血症状。无外伤及手术史。无出血家族史,父母非近亲婚配。家系图见图1A。
家系2:先证者,女,12岁。自幼皮肤易发瘀斑、鼻衄(>10次/年),填塞止血。头部外伤引起颅内出血,输血浆治疗;换牙出血不止(1~3 d),输注血浆止血;自发性右侧大腿肌肉血肿1次。FⅤ∶C<1.0%;无出血家族史,父母非近亲婚配。家系图见图1B。
家系3:先证者,女,43岁。自幼无异常出血史。子宫肌瘤术前筛查发现凝血功能异常,PT、APTT延长,FⅤ∶C<1.0%。父母非近期婚配。家系图见图1C。
家系4:先证者,女,35岁。自幼鼻衄,皮肤易发瘀斑,月经量增多。自然分娩及剖宫产各产1胎;剖宫产术前检查发现凝血功能异常,预防性输注血浆制剂,手术无异常出血,FⅤ∶C为2.5%。父母为非近亲婚配。家系图见图1D。
家系5:先证者,男,41岁。因视力模糊,排除器质性病变后,做凝血相关检查发现APTT、PT均延长,FⅤ∶C为1.6%。自幼有鼻衄。无手术史。父母无异常出血史,非近亲婚配。家系图见图1E。
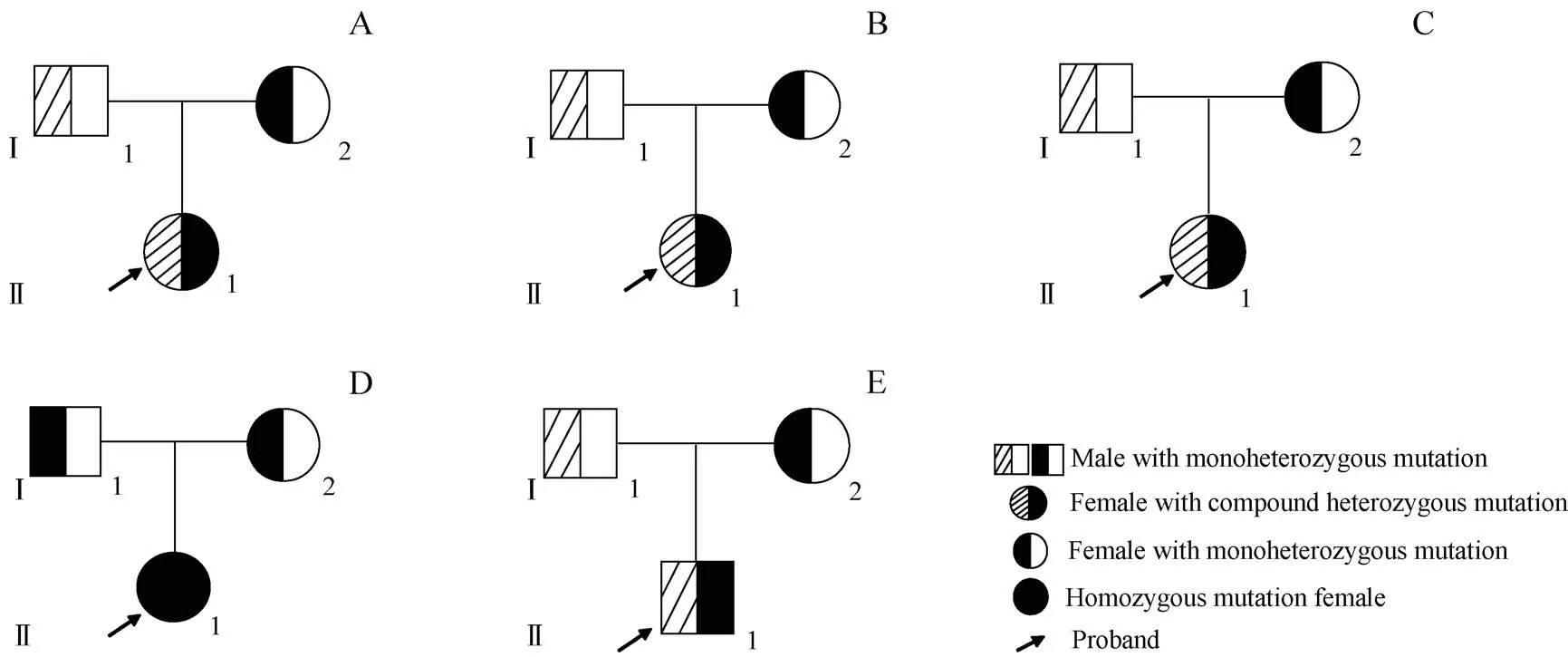
图1 5例FⅤD患者家系图Fig 1 Pedigreediagram of 5 patients with inherited FⅤD
1.2 评分办法
所有先证者的临床出血表现依照ISTH推荐的出血评分问卷进行问诊和记录[8]。该评分系统通过问卷形式对患者出血部位和出血程度进行细致的等级划分,评分较为直观地评估了患者的出血倾向。儿童>2分,成年男性>4分,成年女性>5分,即为有较高的出血风险[9]。
1.3 试验方法
1.3.1 样本采集 采集先证者的外周静脉血标本,用0.109 mol/L枸橼酸钠1∶9抗凝。一部分血液经室温、100×g低速离心10 min后,取上层血浆即富血小板血浆(platelet-rich plasma,PRP)后,于全血细胞计数仪(Beckman Coulter,Inc,美国)上进行血小板计数,用自身乏血小板血浆(platelet-poor plasma,PPP)将血小板数量调至150×109/L,静置数分钟后于2 h内进行TGA。另一部分经2次3 000×g、15min离心后取PPP及血细胞于-80℃冻存待用。血浆用于凝血系统、FⅤ抗原(FⅤ∶Ag)、TFPI检测及凝血酶生成试验,血细胞用于基因提取。
1.3.2 实验室凝血筛查 常规凝血法查PT、APTT。一期凝固法检测FⅤ∶C、FⅧ∶C。仪器及所需试剂购自美国Instrumentation Laboratory公司。
1.3.3 FⅤ∶Ag含量检测 采用双抗体夹心酶联免疫吸附试验(enzyme-linked immunosorbent assay,ELISA)对患者血浆中的FⅤ∶Ag进行检测,仪器及所需试剂购自美国Enzyme Research Laboratories公司。
1.3.4 TFPI抗原检测 生理条件下血浆中存在2种形式的TFPI,即游离型TFPI和脂蛋白结合型TFPI(总TFPI)。其中游离型TFPI仅占总TFPI的20%左右,但主要由游离型TFPI发挥抗凝功能[10]。采用法国Stago公司Assserachrom Total TFPI和Free TFPI试剂盒用ELISA法测定FⅤD患者血浆中总TFPI及游离TFPI水平。
1.3.5 TGA 用自动校正凝血酶曲线法(calibrated automated thrombography,CAT)检测TGA。于96孔板中加入80μL血浆和20μL试剂(样品孔:PPPreagant low/PRPreagant;校准孔:thrombin calibrator)后,放入荧光读数仪内,仪器自动于37℃温育10 min。由仪器自动加入37℃预热的荧光底物,20μL/孔,在激发光390 nm、发射光460 nm的条件下通过检测荧光信号反映待测样本中凝血酶的生成量。96孔板每20 s振荡混匀1次,连续记录1 h内的荧光信号,最后用Thrombinoscope软件处理数据。每份标本分别做2个样本孔及2个校准孔。凝血酶生成曲线主要有4个参数:延迟时间(lagtime,LT)即从反应开始到凝血酶开始生成所经历的时间(min);峰值(peak height)即生成的凝血酶的最大量(nmol/L);达峰时间(time to peak,ttpeak)即从反应开始到凝血酶到达最大量所经历的时间(min);凝血酶生成潜力(endogenous thrombin potential,ETP)即凝血酶生成曲线下的微积分面积,整体反映凝血酶生成的量。其中,指标项峰(peak)和ETP的升高较为特异地反映了促凝功能的增强。TGA反应所需试剂和耗材均购自法国Diagnostica Stago公司。
1.3.6 FⅤ基因(F5)测序和拷贝数变异检测 采用DNA抽提试剂盒(QIAGEN,德国)并严格按照试剂盒说明书所述流程进行操作,抽提先证者及其家系成员外周血基因组DNA。根据F5序列(GenBank NG_011806.1),设计扩增25个外显子及其侧翼序列引物,引物序列及PCR条件参照文献[11]。引物由北京六合华大基因科技有限公司合成。在Thermo Fisher ProFlex™PCR仪上扩增F5的相应片段DNA。PCR产物经1%~2%琼脂糖凝胶电泳观察条带亮度,并送北京六合华大基因科技有限公司测序。用Chromas软件将测序结果与美国NCBI基因文库中的F5序列进行比对(blast),找寻相关致病突变,发现突变位点则通过反向测序予以证实。采用基于竞争性荧光PCR技术为基础的AccuCopy多重基因拷贝数检测技术[12](上海天昊生物科技有限公司)对FⅤD患者F5进行拷贝数变异(copy number variation,CNV)检测。
1.3.7 异位转录水平研究 酚-氯仿法抽提外周血白细胞中的mRNA,使用M-MLV逆转录试剂盒(Thermo Fisher公司,美国)将mRNA逆转录成cDNA。在检测到的大片段缺失和内含子突变的序列前后分别设计PCR引物,对先证者cDNA分别进行扩增。扩增时用正常人外周血cDNA标本作为阳性对照。将2轮扩增产物进行琼脂糖凝胶电泳分析,纯化后与T载连接(TaKaRa,中国),进行TA克隆,分布挑取50个单个菌落测序,分析可能存在的不同的转录本。引物序列见表1。

表1 引物序列信息(5´→3´)Tab1 Primer sequence(5´→3´)
2 结果
2.1 凝血系统检测结果、FⅤ∶Ag及TFPI水平
5例先证者均表现为APTT和PT明显延长,FⅧ∶C均正常;FⅤ∶C和FⅤ∶Ag含量均明显降低,属于Ⅰ型FⅤD,其中有2例先证者的FⅤ∶C和FⅤ∶Ag均<1%,属于重型FⅤD。5例先证者的游离TFPI[(3.54±1.00)ng/mL]和总TFPI[(46.38±12.45)ng/mL]水平有不同程度的降低(表2)。

表2 FVD患者凝血系统、FV∶Ag及TFPI水平检测结果Tab2 Result of coagulation system test,FⅤ∶Ag antigen and TFPIlevel in patients with FⅤD
2.2 TGA结果和出血评分
由于PRP标本需即刻检测,因此只收集到先证者1和2的PRP标本。对其进行检测发现先证者1的TGA-PRP结果和正常对照相比,除了LT和ttpeak稍延长外,凝血酶生成曲线与正常无异。但先证者2的TGA-PRP无法形成凝血酶生成曲线。5例先证者的TGA-PPP结果:LT[(10.43±1.79)min]和ttpeak[(14.33±2.25)min]均明显延长;除1例先证者(先证者2)的peak height极其低和ETP为零且无法形成曲线外,其余4例先证者的peak height[ (184.57±129.36) nmol/L] 和 ETP[(1 200.29±790.03)nmol/(L·min)]均明显高于正常对照。结合先证者2的严重出血表现和TGA-PRP/PPP结果可大致得出患者有较高的出血风险。PRP与PPP结果具有平行性(表3,图2)。4例先证者的出血评分在1~7之间,1例出血评分为16分(表4)。
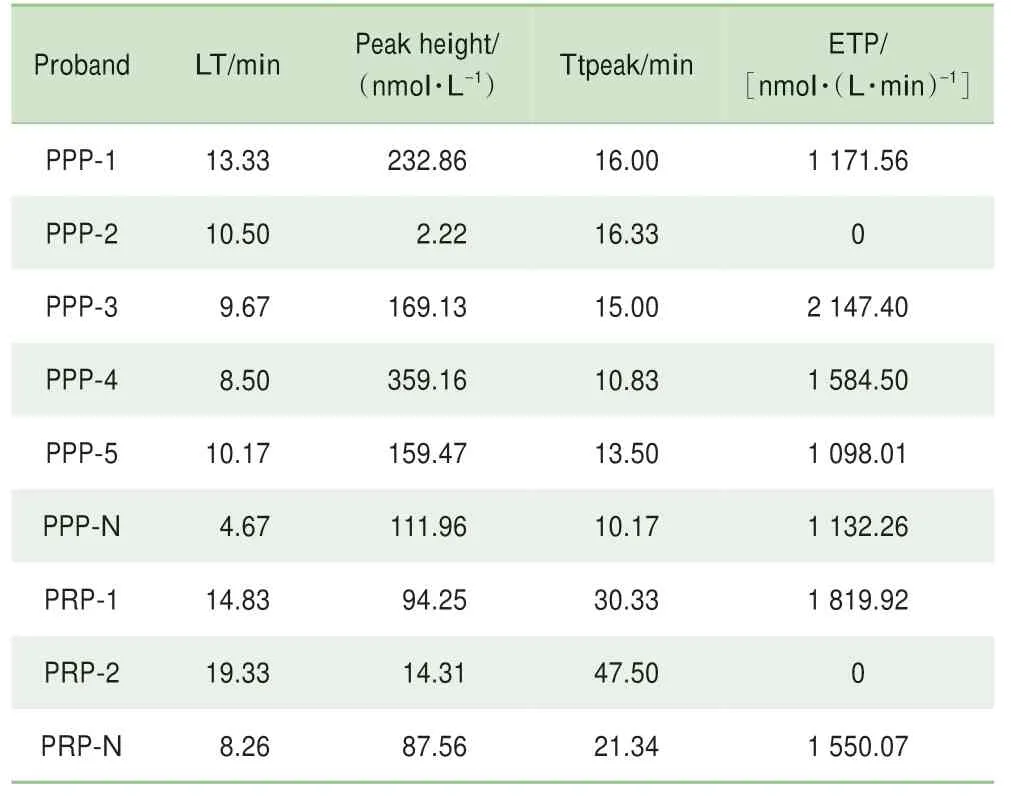
表3 TGA检测结果Tab 3 Results of thrombin generation assay
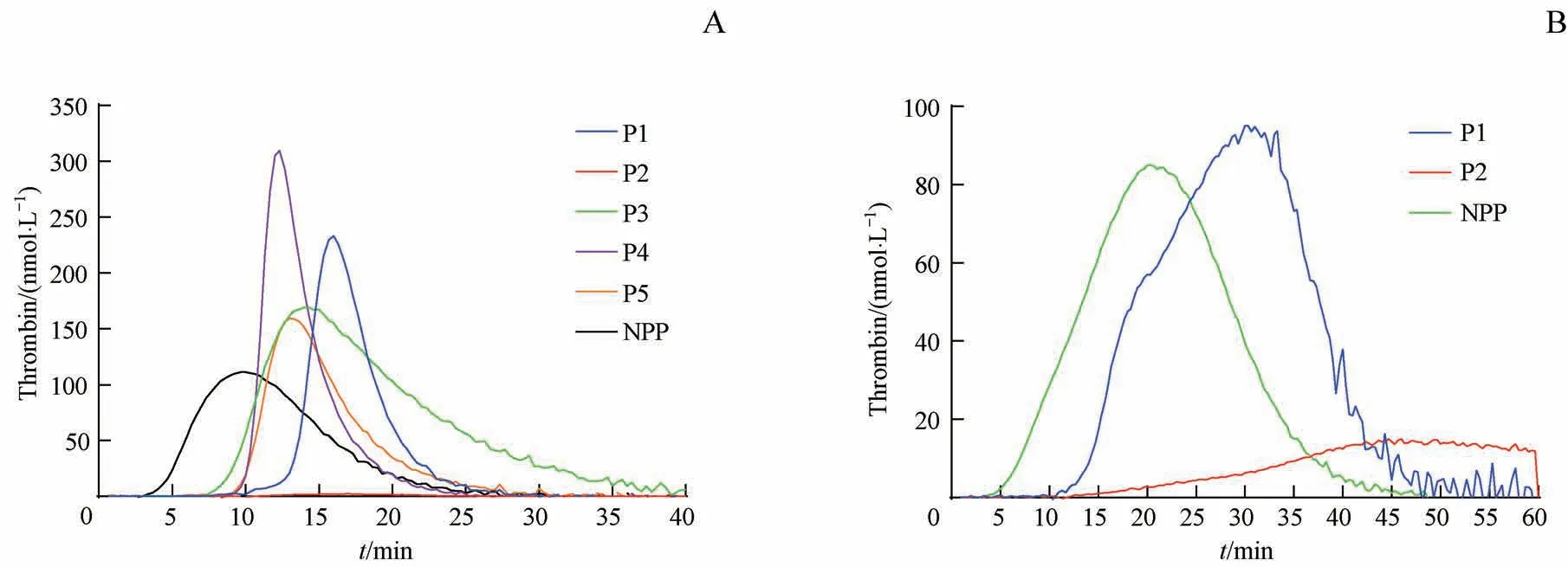
图2 FⅤD患者凝血酶生成试验Fig 2 Thrombin generation assay in patients with FⅤD
2.3 F5、CNV及mRNA水平结果
通过Chromas软件将5例先证者F5测序结果与NCBI基因文库NG_011806.1进行比对(blast),发现8种突变,突变类型有错义突变、无义突变、移码突变、CNV,其中错义突变占75%;p.Cys603Ser、p.Leu949Trpfs*、p.Leu1262_Gln1657del为新突变(表4)。CNV检测发现先证者2的外显子13~14的大片段缺失(c.3784_4971del,p.Leu1262_Gln1657del);mRNA水平进一步分析提示缺失导致患者mRNA剪接异常,产生新的剪切位点,进而表达3种异常转录本——c.3577_4971del、c.3577_4456del、c.3331_4456del(图3)。
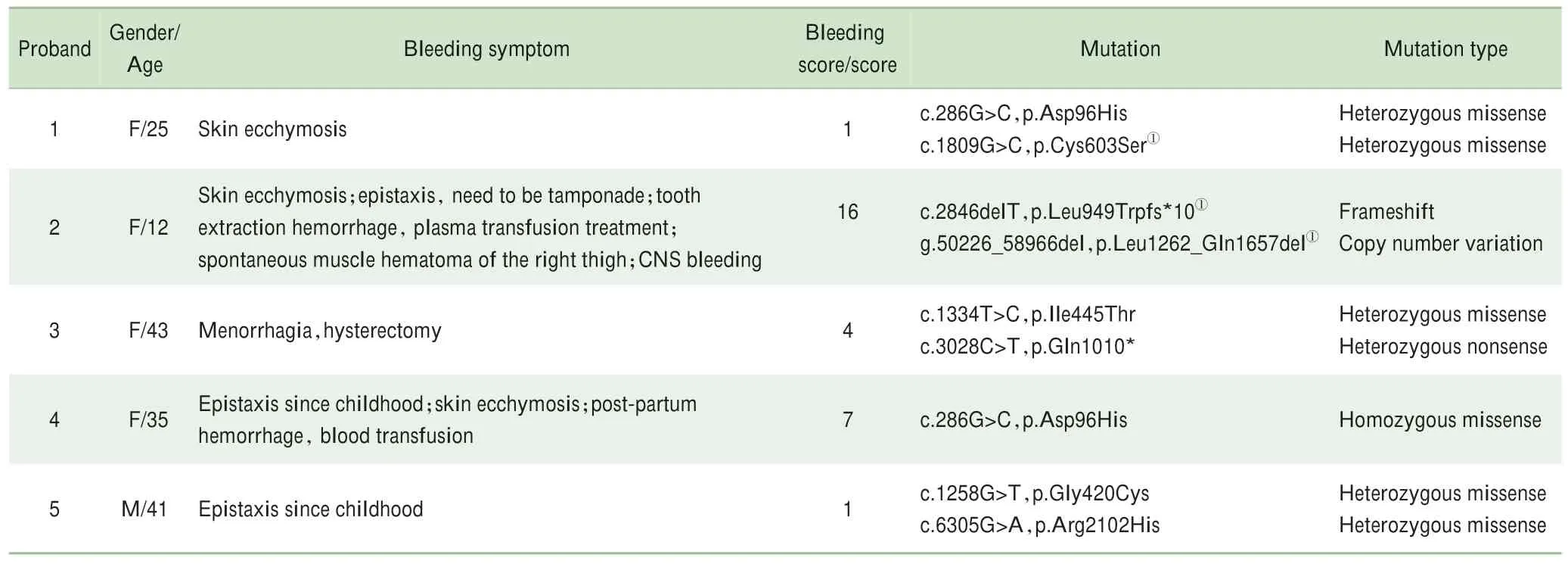
表4 5例遗传性FⅤ缺乏患者表型和基因型Tab 4 Phenotypesand genotypesof patientswith hereditary FⅤdeficiency
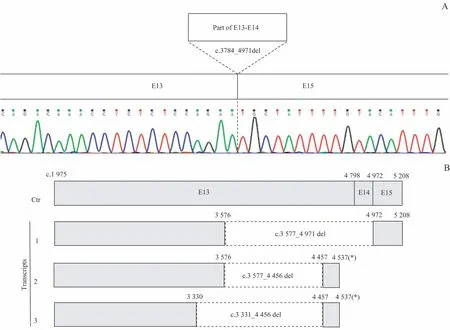
图3 异位转录分析Fig 3 Ectopic transcriptional analysis
3 讨论
FⅤ是调控并参与凝血初始反应的必需因子,其活化形式FⅤa与Ca2+、磷脂和FⅩa形成凝血酶原复合物,是体内凝血酶原最主要的激活物。因而FⅤ缺乏往往导致二期止血功能不全[13],表现为APTT和PT的同时延长。FⅤ为单链糖蛋白,相对分子质量为330 000。成熟FⅤ蛋白结构为A1-A2-B-A3-C1-C2,B结构域在活化过程中被剪切除去。目前报道的FⅤD相关的F5突变约有180种,包括同义突变、错义突变、无义突变、剪切位点突变、插入和缺失等[14]。
本文5例FⅤD患者的APTT和PT均明显延长,FⅤ∶C和FⅤ∶Ag同时降低,属于Ⅰ型FⅤD;有2例先证者的FⅤ活性和抗原水平均<1%,属于重型FⅤD。FⅤD的临床出血表型呈现一定的异质性,轻者无异常出血症状,重者可危及生命,而APTT、PT及凝血因子活性都不能有效地监测患者的临床出血症状。TGA是一个系统监测待测标本中凝血酶生成总潜力的试验[15]。凝血酶在血栓与止血过程中发挥核心作用,运用TGA实时监测凝血酶的含量可以有效地评价出血或血栓形成的风险[16]。本研究中,4例FVD患者的峰值和ETP(TGA-PPP)均高于正常对照(表3和图2),其出血症状也没有预期严重。原因可能为微量的FⅤ已足够产生维持体内最低需量凝血酶。Duckers等[7]研究发现在PPP中,10%FⅤ即可产生正常量的凝血酶。虽然这4例FⅤD患者血浆FⅤ含量均低于10%,但我们推测其血小板中仍含有一定量的FⅤ可以代偿血浆中减少的FⅤ发挥促凝功能。FⅤD患者血浆FⅤ水平和血小板FⅤ水平同步降低,但因为血小板来源的FⅤ比血浆FⅤ具有更强的促凝活性[4],而且血小板在受损部位可以快速释放形成局部高浓度[6],所以血小板FⅤ比血浆FⅤ促凝活性更高,极大地降低了FⅤD患者的出血风险。原因也可能为患者体内低水平的TFPI导致血浆的抗凝功能减弱而产生促凝作用。FⅤ和TFPI在血浆中形成复合物[17],两者水平高度相关,从正常人到单一杂合突变的FⅤD患者和纯合或双杂合突变的FⅤD患者中,血浆中TFPI水平逐渐降低[7]。低水平的TFPI减弱了血浆的抗凝功能,可使FⅤD患者血浆中凝血酶生成的FⅤ最低需量降至1%[18]。
2例FⅤ∶C<1%的重型FⅤD患者(先证者2和3),出血表型和出血评分差异显著。先证者2的出血评分为16分(表4),TGA-PPP和TGA-PRP结果显示均无凝血酶的产生(图2);先证者3的出血评分为4分,TGA-PPP结果的peak height和ETP均明显高于正常对照(图2)。重型FⅤD患者的出血严重程度或与FⅤ∶C水平无关,而与F5突变类型有关。先证者2的F5突变为移码突变p.Leu949Trpfs*10和CNV导致的大片段缺失(表4)。p.Leu949位于FⅤ蛋白的B区,移码突变导致翻译的提前终止,产生FⅤ截短蛋白;FⅤ截短蛋白部分B区缺失,轻链则完全消失,分子链变短。FⅤ结构完整性受到影响,加上氨基酸序列的改变以及催化活性区的部分缺失影响蛋白质的功能,使其表达发生障碍。CNV导致的大片段缺失(c.3784_4971del,p.Leu1262_Gln1657del),在断裂点的上游产生了新的“GT”供位剪接识别位点,进而表达3种异常转录本(c.3577_4971del、c.3577_4456del、c.3331_4456del),c.3577_4971del缺失导致FⅤ蛋白从Val1193至Gln1657缺失465个氨基酸,对B区功能造成一定影响;c.3577_4456del、c.3331_4456del缺失碱基后发生移码突变导致翻译的提前终止,产生FⅤ截短蛋白[19](图3)。产生的截短蛋白或可被体内无义介导的mRNA降解机制(nonsense-mediated mRNA decay,NMD)降解。该患者无凝血酶产生或许与其F5突变类型有关。这2种复合杂合突变是该先证者血浆中几乎没有FⅤ蛋白表达和临床出血表现严重甚至危及生命的原因(表4)。
既往研究[20]发现Asp96His突变(成熟蛋白质为Asp68His)可能是亚洲人的热点突变。研究[21-22]认为Asp68His突变发生后,维持A1区β片层结构的氢键及分子表面范德瓦耳斯力发生偏移,降低了分子结构的稳定性,从而影响FⅤ-Asp68His突变蛋白的折叠、细胞内转运、分泌。在FⅤ蛋白结构中,603Cys与684Cys形成二硫键,这样在A2区就含有一个82个氨基酸组成的β环状结构,此结构对于维持FⅤ的结构及其功能都是极其重要的。杂合错义突变Cys603Ser中,Ser603不能与684Cys形成二硫键,从而导致A2区β环状结构的形成障碍,影响FⅤ的结构稳定性、合成、分泌障碍,进而导致血浆中FⅤ含量下降[13,23]。杂合错义突变Ile445Thr(成熟蛋白质为Ile417Thr),Kling等[24]将其命名为FⅤOgden;FⅤOgden突变导致FⅤ重链A2区氨基端附近的Ile417被Thr替换,用极性亲水氨基酸Thr取代非极性疏水氨基酸Ile417可能足以改变FⅤ蛋白的二级结构,从而对活性产生负面影响,因为表面亲水性氨基酸位置的变化可能会影响FⅤ重链与FXa或凝血酶原的结合,从而影响凝血酶原复合物的形成,进而导致出血症状的发生。杂合错义突变Arg2102His[25](成熟蛋白质为Arg2074His)位于C2区域,FⅤ的晶体结构和H2074的分子C2结构域建模研究表明,FⅤ蛋白的正确折叠需要R2074;Arg2074His影响FⅤC2结构域的结构稳定性和辅因子活性。杂合无义突变p.Gln1010*,基因的无义突变在阅读框中产生提前终止密码子,在细胞中翻译出截短蛋白,可能会对细胞产生毒害作用[26],亦有可能被NMD机制识别并降解。
通过对这5例先证者的基因分析并结合其TGA结果以及出血评分情况发现FⅤD患者的出血严重程度或与F5突变类型有关。轻至中度出血的FVD患者,比如出血评分在1~7之间的4例FⅤD患者,F5突变类型多为错义突变、无义突变[20,27-28];而危及生命的出血,比如出血评分为16分的先证者2,F5突变类型多为剪切位点突变、大片段缺失的重型FⅤD患者[29-32]。这可能是因为无义突变、错义突变等突变类型只影响FⅤ蛋白的局部功能,比如FⅤ蛋白的折叠、细胞内转运、分泌等,对其促凝结构域的影响不大。但大片段缺失、剪切位点突变这些突变类型会导致FⅤ蛋白的严重缺失,进而导致严重的出血。
由于FⅤ在凝血过程中发挥重要作用,对其分子机制及其功能的结构基础研究具有重要意义。尤其在临床上,对FⅤD患者进行常规基因检测,根据其基因突变类型对出血严重程度的判断具有一定意义。
参·考·文·献
[1] Duckers C,Simioni P,Rosing J,et al.Advances in understanding the bleeding diathesis in factorⅤdeficiency[J].Br J Haematol,2009,146(1):17-26.
[2] Lippi G,Favaloro EJ,Montagnana M,et al.Inherited and acquired factorⅤdeficiency[J].Blood Coagulation Fibrinolysis,2011,22(3):160-166.
[3] Dahlbäck B.Pro-and anticoagulant properties of factorⅤin pathogenesis of thrombosis and bleeding disorders[J].Int JLab Hematol,2016,38(Suppl 1):4-11.
[4] Tabibian S,Shiravand Y,Shams M,et al.A comprehensive overview of coagulation factorⅤand congenital factorⅤdeficiency[J].Semin Thromb Hemost,2019,45(5):523-543.
[5] MonkovićDD,Tracy PB.Functional characterization of human plateletreleased factorⅤand its activation by factor Xa and thrombin[J].J Biol Chem,1990,265(28):17132-17140.
[6] Gremmel T,Frelinger AL,Michelson AD.Platelet physiology[J].Semin Thromb Hemostasis,2016,42(3):191-204.
[7] Duckers C,Simioni P,Spiezia L,et al.Low plasma levels of tissue factor pathway inhibitor in patients with congenital factorⅤdeficiency[J].Blood,2008,112(9):3615-3623.
[8] Rodeghiero F,Tosetto A,Abshire T,et al.ISTH/SSC bleeding assessment tool:a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders[J].J Thromb Haemost,2010,8(9):2063-2065.
[9] Elbatarny M,Mollah S,Grabell J,et al.Normal range of bleeding scores for the ISTH-BAT:adult and pediatric data from the merging project[J].Haemophilia,2014,20(6):831-835.
[10] Bridey F,Lacombe C,Sustendal L,et al.Development of a method to separate lipoprotein-bound and lipoprotein-depleted tissue factor pathway inhibitor.Measurement of free tissue factor pathway inhibitor activity[J].Blood Coagul Fibrinolysis,1998,9(7):637-643.
[11] Fu QH,Zhou RF,Liu LG,et al.Identification of three F5 gene mutations associated with inherited coagulation factorⅤdeficiency in two Chinese pedigrees[J].Haemophilia,2004,10(3):264-270.
[12] Du RQ,Lu CC,Jiang ZW,et al.Efficient typing of copy number variations in a segmental duplication-mediated rearrangement hotspot using multiplex competitive amplification[J].JHum Genet,2012,57(8):545-551.
[13] Duga S,Asselta R,Tenchini ML.Coagulation factorⅤ[J].Int JBiochem Cell Biol,2004,36(8):1393-1399.
[14] Al-Numair NS,Ramzan K,Saleh M,et al.First description of the molecular and clinical characterization of hereditary factor V deficiency in Saudi Arabia:report of four novel mutations[J].Blood Coagul Fibrinolysis,2019,30(5):224-232.
[15] 黄丹丹,陆晔玲,戴菁,等.凝血酶生成试验在遗传性凝血因子缺陷症患者出血评估中的价值[J].中国实验诊断学,2011,15(3):503-507.
[16] Tripodi A.Thrombin generation assay and its application in the clinical laboratory[J].Clin Chem,2016,62(5):699-707.
[17] van Doorn P,Rosing J,Duckers C,et al.FactorⅤhas anticoagulant activity in plasmain thepresenceof TFPIα:differencebetween FV1and FV2[J].Thromb Haemost,2018,118(7):1194-1202.
[18] Shao YY,Wu WM,Xu GQ,et al.Low factor V level ameliorates bleeding diathesis in patients with combined deficiency of factorⅤand factorⅧ[J].Blood,2019,134(20):1745-1754.
[19] 游国岭,陆晔玲,戴菁,等.F8基因大片段缺失所导致的一例重型血友病A的分子发病机制研究[J].中华检验医学杂志,2013,36(6):534-537.
[20] Park CH,Park MS,Lee KO,et al.Congenital factorⅤdeficiency from compound heterozygous mutations with a novel variant c.2426del(p.Pro809Hisfs*2)in theF5gene:a case report[J].Medicine(Baltimore),2020,99(5):e18947.
[21] Liu HC,Shen MC,Eng HL,et al.Asp68His mutation in the A1 domain of human factorⅤcauses impaired secretion and ineffective translocation[J].Haemophilia,2014,20(4):e318-e326.
[22] 梁枫萍,程鹏.1例遗传性凝血因子Ⅴ缺乏症家系的基因分析[J].重庆医学,2018,47(14):1970-1974.
[23] Mann KG,Kalafatis M.FactorⅤ:acombination of dr Jekyll and mr Hyde[J].Blood,2003,101(1):20-30.
[24] Kling SJ,Griffee M,Flanders MM,et al.FactorⅤdeficiency caused by a novel missense mutation,Ile417Thr,in the A2 domain[J].J Thromb Haemost,2006,4(2):481-483.
[25] Schrijver I,Houissa-Kastally R,Jones CD,et al.Novel factorⅤC2-domain mutation(R2074H)in two families with factor V deficiency and bleeding[J].Thromb Haemost,2002,87(2):294-299.
[26] 傅启华,王鸿利,王明山,等.2种新的凝血因子Ⅴ基因突变导致的遗传性凝血因子Ⅴ缺乏症[J].中华医学杂志,2003,83(4):312-315.
[27] Montefusco MC,Duga S,Asselta R,et al.A novel two base pair deletion in the factorⅤgene associated with severe factorⅤdeficiency[J].Br JHaematol,2000,111(4):1240-1246.
[28] Borhany M,Ranc A,Fretigny M,et al.Molecular analysis of eight severe FⅤ-deficient patients in Pakistan:a large series of homozygous for frameshift mutations[J].Haemophilia,2019,25(4):e278-e281.
[29] Castoldi E,Duckers C,Radu C,et al.Homozygous F5 deep-intronic splicing mutation resulting in severe factorⅤdeficiency and undetectable thrombin generation in platelet-rich plasma[J].JThromb Haemost,2011,9(5):959-968.
[30] Nuzzo F,Beshlawi I,Wali Y,et al.High incidence of intracranial bleeding in factor V-deficient patients with homozygousF5splicing mutations[J].Br JHaematol,2017,179(1):163-166.
[31] Nuzzo F,Bulato C,Nielsen BI,et al.Characterization of an apparently synonymousF5mutation causing aberrant splicing and factorⅤdeficiency[J].Haemophilia,2015,21(2):241-248.
[32] Hunt RC,Simhadri VL,Iandoli M,et al.Exposing synonymous mutations[J].Trends Genet,2014,30(7):308-321.
