基于CRISPR/Cas9技术构建Atg7基因敲除的人脑微血管内皮细胞系
2021-09-06李雯卉刘辉李泽烨韦佳祎陈誉华
李雯卉, 刘辉, 李泽烨, 韦佳祎, 陈誉华
血脑屏障是中枢神经系统的一个独特结构,它紧密地控制着离子、分子、细胞在血管和脑之间运动[1]。血脑屏障的屏障特性是使神经组织免受毒素和病原体的侵袭,对中枢神经系统稳态的维持至关重要[1-2]。脑微血管内皮细胞是血脑屏障重要的细胞组分,它在血脑屏障的建立以及功能发挥中起关键作用[3-4]。许多研究还发现脑微血管内皮细胞的功能障碍与很多脑血管相关疾病有紧密的联系[5-9]。1992年,Atg7因其在酵母自噬中发挥重要作用而开始被关注[10],随后它一些新的功能也陆续被研究者发现。近来有研究发现小鼠内皮细胞敲除Atg7能减弱动脉血栓的形成[11],我们的前期研究也发现,小鼠内皮细胞敲除Atg7不利于脑血管的形成[12]。然而,Atg7在脑血管和参与血脑屏障形态建成的机制尚不清楚。CRISPR/Cas9系统是细菌和古细菌形成的一种免疫防御系统,近年来经人为改造后广泛应用于基因编辑,其原理是通过单一引导RNA序列(sgRNA)识别特定靶DNA序列,将内切酶Cas9引导至靶DNA的设定位置进行切割,使DNA双链断裂,细胞在通过自身非同源性末端修复时,可使目标区域DNA出现插入、缺失等改变,达到基因敲除或改造的目的[13]。我们利用CRISPR/Cas9技术在体外构建Atg7基因敲除的人脑微血管内皮细胞,为Atg7在脑血管的发育和参与血脑屏障形态建成的机制以及脑血管相关疾病的深入研究提供体外细胞模型。
1 材料与方法
1.1 主要实验材料 人脑微血管内皮细胞(human brain microvascular endothelial cells,HBMEC)(美国Johns Hopkins大学医学院K.S.Kim教授赠送);1640培养液、胎牛血清、胰酶、嘌呤霉素(Gbicol,美国);NU血清(BD,美国);杀稻瘟菌素、atg7抗体(Sigma,美国);H5450 pLenti-CMV-Puro-P2A-3Flag-espCas9_1.1;H6804 pLenti-U6-spgRNAv2.0-CMV-Blasticidin质粒;PrimeSTAR®HS DNA Polymerase;TaKaRa TaqTMVer. 2.0 plus dye;TaKaRa MiniBEST Agarose Gel DNA Extraction Kit Ver.4.0;和元无缝克隆试剂盒;DH5α感受态细胞;luria-bertani(LB)培养基;AxyPrep质粒DNA小量试剂盒;无内毒素质粒大提试剂盒(天根生化科技有限公司,DP117,北京);TRNzol Universal总RNA提取试剂(天根生化科技有限公司,DP424,北京);FastKing RT Kit(With gDNase)(天根生化科技有限公司,KR116,北京);荧光定量检测试剂盒(SYBR Green)(天根生化科技有限公司,FP209,北京);放射免疫沉淀测定细胞裂解液(碧云天,美国);二喹啉甲酸(bicinchoninic acid,BCA)蛋白定量试剂盒、冷冻高速离心机(Thermo Fisher,美国);β-actin单克隆抗体(中杉金桥,中国);山羊抗鼠HRP酶标二抗(赛默飞世尔,中国);DNA电泳槽、稳压电泳仪(上海天能科技有限公司);凝胶成像分析仪(培清科技);超微量分光光度计(北京奥凯科技发展有限公司);移液器(Eppendorf,德国);电热恒温水槽、隔水式恒温培养箱、恒温振荡器(上海一恒科学仪器有限公司),聚合酶链式反应(polymerase chain reaction,PCR)仪(Applied biosystems,美国);7500 Real Time PCR扩增仪(ABI,美国);双光子激光共聚焦显微镜(ZEISS,德国)。本研究的引物合成,sgRNA的合成以及病毒包装均由和元生物技术股份有限公司完成。
1.2 sgRNA的设计和合成 利用CRISPR在线设计工具(http://crispr.mit.edu),在人Atg7基因(GeneID:10533)的第一个外显子设计2对sgRNA(表1),分别为sgRNA1(针对的靶点序列AGA AGA AGC TGA ACG AGT ATC GG)、sgRNA2(针对的靶点序列TAG GGT CCA TAC ATT CAC TGA GG);分别在sgRNA的正义链(F)的5’端添加ACCG、反义链(R)的5’端添加AAAC形成黏性末端,可与BsmBI酶酶切后的黏性末端互补。

表1 CRISPR/Cas9敲除HBMEC Atg7基因sgRNA引物序列
1.3 重组真核表达载体H6804-sgRNA的构建 使用限制性内切酶BasmBI对表达载体H6804进行酶切,酶切反应体系为:质粒2 μg,10×反应Buffer 5 μL,限制性内切酶1 μL,用水补足50 μL,于37 ℃水浴锅中孵育2 h以上。酶切产物进行琼脂糖凝胶电泳检测酶切效果,从琼脂糖凝胶电泳切割目的载体条带,用TaKaRa MiniBEST Agarose Gel DNA Extraction Kit Ver.3.0做胶回收,得到质粒H6804的线性载体。使用无缝克隆试剂盒将sgRNA与H6804的线性载体连接,构建重组真核表达载体H6840-sgRNA1(Y7541)和H6840-sgRNA2(Y7542),分别将它们转化到DH5α感受态细胞,将感受态细胞均匀涂到含氨苄青霉素的LB琼脂培养基平板上,37 ℃恒温培养箱中过夜培养,第二天挑取单菌落,摇菌培养后进行菌落PCR,菌落鉴定得到的阳性克隆,送测序公司进行测序验证,正向测序引物hU6-F2 TAC GAT ACA AGG CTG TTA GAG AG,反向测序引物pY-SEQR CTA TTA ATA ACT AAT GCA TGG C。软件比对测序结果并分析。测序通过的阳性克隆,安排质粒小提并进行双酶切再一次验证。
1.4 HBMEC的培养以及嘌呤霉素及杀稻瘟菌素筛选浓度的确定 用NU培养基(含10%NU血清、10%胎牛血清和细胞生长所需营养成分的1640培养液)培养细胞。取处于对数生长期的细胞接种于96孔板,每孔接种1.5×105的细胞量,第二天分别用嘌呤霉素浓度为0、1、2、3、4、5、6、7、8、9、10、20 mg/L的NU培养基或杀稻瘟菌素浓度为0、1、2、3、4、5、6、7、8、9、10、20 mg/L的NU培养基替换旧的培养基。每个抗生素的每个浓度都设置3个复孔,总共72个孔。每天观察细胞的生长情况,选择7 d使细胞全部死亡的最小浓度作为它们的筛选浓度。最后确定嘌呤霉素的筛选浓度为1 mg/L,杀稻瘟菌素的筛选浓度为3 mg/L。
1.5 慢病毒包装和感染HBMEC以及敲除Atg7基因的HBMEC的单克隆筛选 H5450 pLenti-CMV-Puro-P2A-3Flag-espCas9_1.1、H6840-sgRNA1(Y7541)和H6840-sgRNA2(Y7542)三个真核表达载体分别和包装质粒共转染293T细胞,在293T细胞中进行慢病毒包装。之后对包装好的病毒进行滴度检测。培养HBMEC,取对数生长期的细胞接种于2个35 mm培养皿,每个皿接种5×105的细胞量,第二天其中一个培养皿加入Cas9病毒,所加病毒量根据病毒滴度、细胞量和选择的感染复数(multiplicity of infection,MOI)值计算[病毒量(μL)=MOI×感染时的细胞数/滴度(TU/mL)×1 000]。病毒感染12~16 h后更换为新的NU培养基。另一个培养皿不加病毒,作为加Cas9病毒的对照组。待细胞长满后进行细胞传代,Cas9病毒感染的细胞传至4个35 mm培养皿,分别编号为1、2、3、4,用含1 mg/L嘌呤霉素的NU培养基培养。未感染Cas9病毒的正常HBMEC统一保留一个35 mm培养皿,编号为5,用正常的NU培养基培养。4号和5号培养皿中细胞长满之后,用来提取蛋白质,通过蛋白免疫印迹实验检测Cas9。1号和2号培养皿中分别加入H6840-sgRNA1(Y7541)和H6840-sgRNA2(Y7542)病毒感染细胞,感染过程与Cas9病毒感染细胞相同。感染48 h后消化离心收集细胞,重悬细胞计数,通过有限稀释法最后把细胞接种于96孔板,每孔接种1个细胞,各接种48个孔,进行单克隆筛选。接种96孔板后剩下的细胞传至60 mm培养皿继续培养,此时的培养液用含1 mg/L的嘌呤霉素和3 mg/L的杀稻瘟菌素的NU培养基,待细胞长满后冻存备用。96孔板中细胞第二天也换成1 mg/L的嘌呤霉素和3 mg/L的杀稻瘟菌素的NU培养基。每天观察孔中细胞的生长情况,并做好标记。培养5 d后每隔1天对有细胞的孔进行换液,大约14 d后待孔中细胞长满之后,转移至24孔板中,继续扩大培养后依次传至6孔板、培养瓶中。对挑选出来的单克隆细胞都仔细做好标号。3号培养皿中的细胞(只感染了Cas9病毒的细胞,HBMEC-cas9)继续扩大培养,冻存一部分,其作为我们挑选出来的单克隆细胞(HBMEC-atg7KO)的对照细胞。
1.6 HBMEC-Cas9和HBMEC-atg7KO中Atg7 mRNA的检测 采用实时荧光定量PCR检测Atg7 mRNA的相对表达量。收集HBMEC-Cas9和HBMEC-atg7KO,通过TRNzol Universal总RNA提取试剂提取细胞的总RNA,逆转录合成cDNA,以cDNA为模板进行实时荧光定量PCR扩增,检测细胞的Atg7 mRNA。引物序列见表2。反应条件为:95 ℃预变性15 min,95 ℃变性10 s,60 ℃退火延伸32 s,扩增40个循环。以GAPDH为内参,根据2-△△Ct计算Atg7的mRNA相对表达量。△△Ct=(CT实验组目的基因-CT实验组内参基因)-(CT对照组目的基因-CT对照组内参基因)。

表2 人Atg7基因和GAPDH基因引物序列
1.7 蛋白免疫印迹实验检测HBMEC-Cas9和HBMEC-atg7KO中Atg7蛋白 采用蛋白免疫印迹实验检测Atg7蛋白,以β-actin作为内参。收集HBMEC-Cas9和HBMEC-atg7KO,磷酸盐缓冲液(phosphate buffered saline,PBS)洗涤2次,用含苯甲基磺酰氟蛋白酶抑制剂的放射免疫沉淀测定细胞裂解液在冰上裂解细胞,裂解30 min后用细胞刮收集到离心管中,4 ℃离心机12 000 r/min离心15 min(离心半径20 cm),上清液即为提取的蛋白质。通过BCA蛋白定量试剂盒对蛋白进行定量,加入5×蛋白上样缓冲液制成蛋白样,制成的蛋白样99 ℃加热5 min。之后即可进行电泳,转膜,5%脱脂奶粉封闭1 h;加一抗(Atg7 1∶1 000;β-actin 1∶2 000)4 ℃孵育过夜,第二天Tris-HCl缓冲盐溶液洗膜3×10 min,二抗(1∶8 000)室温孵育1 h,Tris-HCl缓冲盐溶液洗膜3×10 min,化学发光,观察蛋白的表达情况。
1.8 细胞免疫荧光染色检测HBMEC-Cas9和HBMEC-atg7KO中Atg7蛋白 分别将HBMEC-Cas9和HBMEC-atg7KO接种于孔中已提前放好盖玻片的6孔板中,各孔接种2×105的细胞量,细胞培养箱中培养1 d后取出细胞进行染色。去除培养基,用PBS清洗2次,加入4%多聚甲醛室温固定10 min;固定后用预冷的PBS摇床上清洗3次,每次5 min;用0.1%的Triton X-100室温透化处理1 min,预冷的PBS摇床上清洗3次,每次5 min;加入1%的牛血清白蛋白室温封闭1 h;封闭结束后加入鼠抗人Atg7一抗(1∶100,用1%牛血清白蛋白稀释),4 ℃孵育过夜;第二天用预冷的PBS摇床上清洗3次,每次5 min;加入Alexa Fluor 488标记的山羊抗鼠二抗(1∶200,用1%牛血清白蛋白稀释),室温孵育1 h;1 h后用预冷的PBS摇床上清洗3次,每次5 min,之后进行细胞核染色(1 g/L的4’6-二脒基-2-苯基吲哚(4'6-diamidino-2-phenylindole,DAPI)用PBS按1∶1 000稀释),染色3 min;预冷的PBS摇床上清洗3次,每次5 min;加入防淬灭的封固剂进行封固,共聚焦显微镜照相观察。
1.9 显微镜下观察细胞形态 在35 mm培养皿中培养正常HBMEC、HBMEC-Cas9和HBMEC-atg7KO,在倒置显微镜下观察3种细胞的形态,并拍照记录,分析敲除Atg7后细胞形态是否出现变化。

2 结果
2.1 重组真核表达载体H6804-sgRNA的构建情况 各挑取8个含重组真核表达载体H6804-sgRNA1(Y7541)或H6804-sgRNA2(Y7542)的单菌落摇菌后进行菌落PCR扩增,均可见清晰条带。同时基因测序结果发现,重组真核表达载体H6804-sgRNA1相对质粒H6840插入了一段序列,sgRNA1-F序列一致;重组真核表达载体H6804-sgRNA2相对质粒H6840插入了一段序列,与sgRNA2-R序列一致。以上结果表明,重组真核表达载体H6804-sgRNA1、H6804-sgRNA2构建成功。见图1。
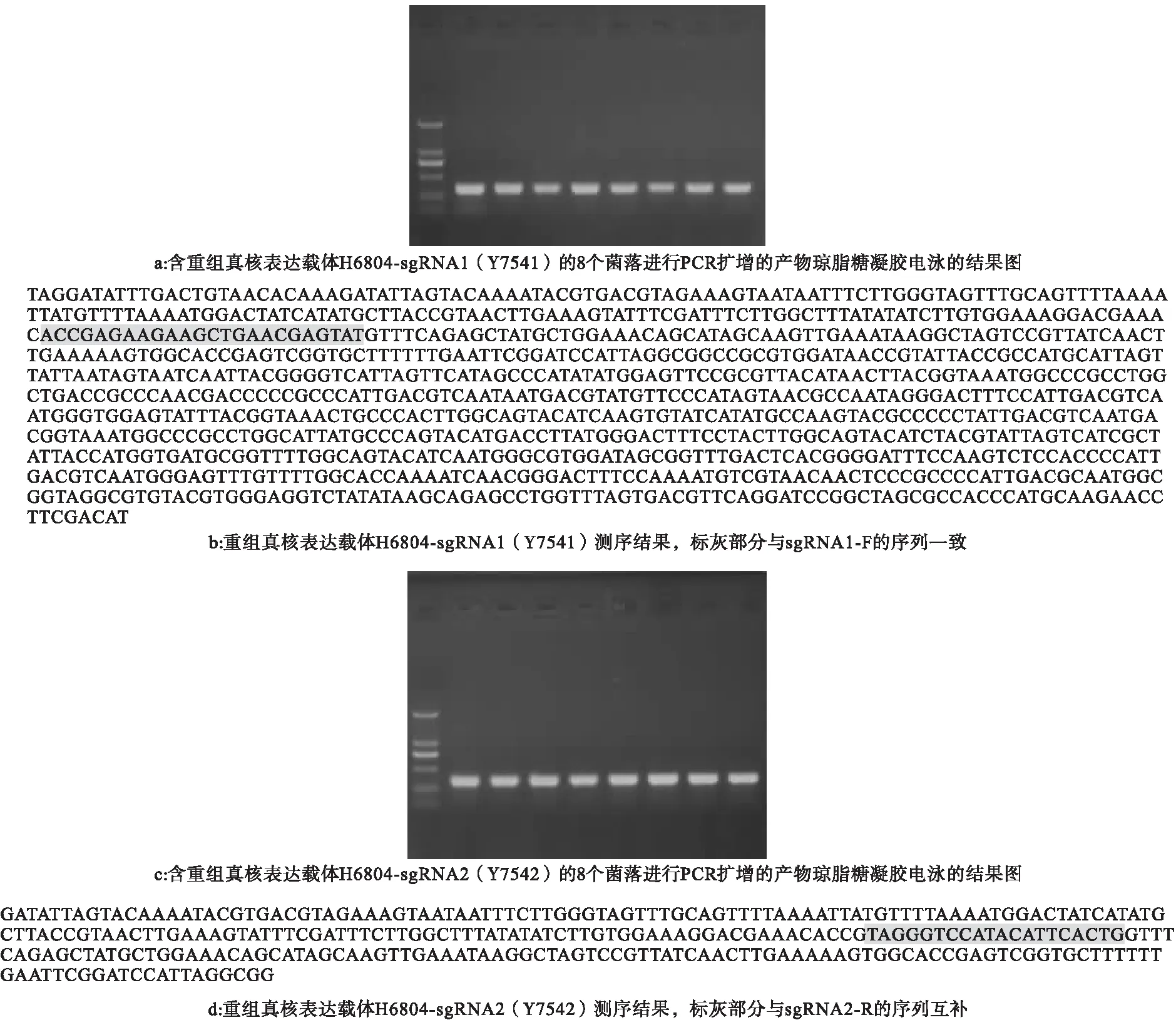
图1 重组真核表达载体H6804-sgRNA的构建
2.2 实时荧光定量PCR检测HBMEC-Cas9和HBMEC-atg7KO中Atg7 mRNA的相对表达 通过实时荧光定量PCR实验检测Atg7的mRNA,结果显示,Atg7基因敲除的HBMEC Atg7 mRNA的表达量明显低于对照组,差异有统计学意义(P<0.05),见图2。

图2 HBMEC-Cas9和HBMEC-atg7KO中Atg7 mRNA的相对表达
2.3 蛋白免疫印迹检测HBMEC-Cas9和HBMEC-atg7KO中Atg7蛋白的表达 通过蛋白免疫印迹实验,我们发现CRISPR/Cas9技术构建的Atg7基因敲除的HBMEC Atg7蛋白的表达完全消失。见图3。

图3 HBMEC-Cas9和HBMEC-atg7KO中Atg7蛋白的检测
2.4 细胞免疫荧光检测HBMEC-Cas9和HBMEC-atg7KO中Atg7蛋白的表达 利用免疫荧光染色观察HBMEC-Cas9和HBMEC-atg7KO中Atg7蛋白,结果表明HBMEC-atg7KO组与对照组HBMEC-Cas9相比Atg7的表达明显减少。见图4。

图4 免疫荧光染色观察Atg7的表达
2.5 光镜下HBMEC-Cas9和HBMEC-atg7KO的细胞形态 培养HBMEC-Cas9和HBMEC-atg7KO细胞,在光镜下观察它们的形态,Atg7敲除组与对照组相比,未见有明显的细胞形态改变。见图5。

图5 细胞形态的观察(×10)
3 讨论
CRISPR/Cas9系统在1987年由Ishino等[14]发现以来,已经成为生物学家和遗传学家最强大的工具之一。从2012年CRISPR/Cas9首次证明可以在体外进行DNA切割以来,该系统因为其比第一代ZFN和第二代TALEN基因编辑技术操作更为简单、修饰更为精确、效率更为高效、成本更低等优点而广泛应用[15]。该系统通过gRNA和Cas9蛋白可实现对靶基因DNA的识别和编辑;除了对单个碱基编辑外,我们可设计多个不同的gRNA介导Cas9蛋白同时进行多基因的编辑。
自噬中的关键基因Atg7作为研究的焦点,在发育、维系健康和控制疾病中发挥重要作用[16]。在前期研究中,在体实验我们构建了内皮特异性敲除Atg7的小鼠;而体外实验我们只能选择RNAi技术瞬时敲减Atg7。由于RNAi技术只能短时间内使Atg7基因的表达下调,这极大的限制了我们对该基因功能的研究,而CRISPR/Cas9技术的应用使得这一问题迎刃而解。本研究中我们针对人Atg7基因第一个外显子设计了2对sgRNA,构建重组质粒,与Cas9质粒共转到HBMEC中,实现对HBMEC细胞进行Atg7基因的敲除。通过实时荧光定量PCR、蛋白免疫印迹实验和细胞免疫荧光染色证明了我们在HBMEC细胞中成功敲除了Atg7基因。通过在光镜下对Atg7敲除的HBMEC和对照组的细胞进行细胞形态观察,我们未发现形态上有明显改变。
综上所述,我们利用CRISPR/Cas9技术成功构建了Atg7基因稳定敲除的HBMEC,这将极大促进我们对Atg7基因在脑血管的发育和血脑屏障形态建成的机制以及脑血管相关疾病的研究。
