1例Rubinstein-Taybi综合征的临床分析及文献复习
2021-09-06许金波童光磊沈瑞丽王羽辰
许金波, 童光磊, 沈瑞丽, 王羽辰
Rubinstein-Taybi综合征(Rubinstein-Taybi syndrome,RSTS),又称宽拇指巨趾综合征、巨指(趾)综合征,是一类罕见的常染色体显性遗传性疾病。该病的临床特征为运动发育迟缓、智能障碍、拇指或脚趾宽大、特殊面容、头小畸形、脏器畸形等。Rubinstein和Taybi 1963年首次报道了该罕见病RSTS[1]。该病在新生儿中的发病率为1∶100 000~1∶125 000[2]。目前国外已报道260余例,国内报道10余例。现阶段对RSTS的诊断主要结合临床特征及基因检测技术,其中55%~70%的病例通过基因检测得以确诊[3]。RSTS作为一种罕见病,临床容易误诊漏诊,本研究对1例RSTS患儿采用全外显子测序技术,确认存在CREBBP基因的新生突变,现将其临床资料总结报告如下。
1 临床资料
1.1 一般情况 患儿女,2岁,因“出生至今2岁不能独站独走,主动表达少”入院。患儿出生后家人逐渐发现其运动发育及言语发育落后于正常同龄儿童,8月龄翻身,1岁余仍不能独坐,至我院行综合康复治疗后渐能独坐,现2岁余仍不能独站行,扶站时双足尖足,言语仅能无意识发“baba”“mama”等叠音,病情呈非进行性进展。患儿系足月自然娩出,出生时无缺氧、黄疸等病史。母孕期未见明显异常。喂养史及疫苗接种史均正常。患儿父母均体健,否认慢性疾病史,无家族遗传性疾病史。
体格检查:身高70 cm(低于正常同龄女童参考值第三百分位点),体质量12 kg(位于正常同龄女童参考值第50~75百分位点),头围43 cm(低于正常同龄女童参考值第三百分位点),前囟已闭,眉毛浓密,眉弓高,双眼距宽,颈软,无抵抗,心肺腹检查未见明显异常,右手通贯掌,双手拇指及第一脚趾粗大(见图1),四肢肌力4级,双下肢胫前肌肌张力增高,双膝反射(++),双巴氏征(-)。粗大运动功能发育落后:抬头可,独坐可,不能独站,不能独行,不能双脚跳;精细动作发育落后:能将玩具从一手换入另一手,拇指、示指对指拿东西动作笨拙,不会用勺子吃饭;言语发育落后:能发“baba”“mama”等复音,但无意识,不能说单字或词,不能指出自己的手、眼。
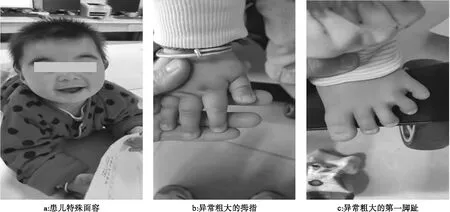
图1 患儿面部及手足部特征
1.2 辅助检查 患儿血常规、尿常规、大便常规、肝肾功能、心肌酶谱、电解质、甲状腺功能、染色体计数及核型分析、新生儿遗传代谢病筛查均未见明显异常。脑干听觉诱发电位:双耳反应阈55 db,右侧A波、B波、R波潜伏期延长,Ⅲ~Ⅴ、Ⅰ~Ⅴ波峰间值较对侧延长。脑电图:正常范围儿童脑电图。发育商量表测评:实测发育龄5.9月龄。
头颅磁共振平扫检查提示双侧大脑半球白质髓鞘化发育异常,侧脑室前后脚锐利,胼胝体短薄。见图2。

图2 患儿头颅磁共振平扫检查影像
1.3 基因检测 患儿行多个疗程康复治疗后,症状未见明显改善,为进一步探寻病因,了解预后,经患儿家属知情同意后,分别抽取患儿及父母亲外周血3 mL,进行全外显子测序分析。全外显子组测序采用IDT The xGen Exome Research Panel v1.0全外显子捕获芯片,经Illumina NovaSeq 6000系列测序仪(PE150)测序完成,且目标序列测序覆盖度不低于99%。经过集分子生物学注释、生物学、遗传学及临床特征分析为一体的遗传病精准诊断云平台系统分析筛选,结合致病突变数据库、正常人基因组数据库、已知四千种遗传病临床特征数据库等及基因数据分析算法,对数十万个基因变异进行分级,变异分级采用三要素分级体系以及ACMG(美国医学遗传学会)基因变异分级体系。对目标序列进行聚合酶链反应后,经ABI3730测序仪进行Sanger测序验证,并经序列分析软件得到验证结果。
2 结果
(1)CREBBP基因存在碱基缺失c.3469_3471del,p.val1157del(杂合),父母该位点为正常基因型,提示该变异可能为新生突变;(2)其他基因(目前临床症状相关),未发现病理性变异。该例样本中变异c.3469_3471del,p.val1157del为新发现的变异,Alamut功能软件预测可能会影响蛋白结构域的功能,按照ACMG变异分类标准,可以归纳为“可能致病性likely pathogenic”变异。
3 讨论
RSTS作为一种罕见病,临床报道较少,尚无明确统一的诊断标准及治疗方案。RSTS在胚胎期生长发育多正常,出生时或婴儿期经常被发现,其面部及手足特征鲜明[4]。RSTS的特殊面容有拱形粗眉、眼距增宽、宽鼻梁、喙状鼻、高弓上颚、小颌畸形等[5]。该例患儿除了粗大的拇指及第一脚趾这一特征性表现外,还可见眉毛粗黑、眼距增宽、通贯掌等发育畸形。此外,该例患儿还表现为身高、体质量等发育指标延迟,粗大运动功能及言语功能发育落后,现2岁仍不能独站行,且扶站扶行时姿势异常,言语只能无意识发“baba”“mama”音,明显落后于正常同龄儿童。运动发育迟缓和语言发育落后是大多数罕见病首诊的主要原因。临床发现约21%的RSTS患儿在生后数月出现生长发育迟缓,常低于同龄儿童的第5百分位以下[6]。RSTS患儿均有不同程度的认知障碍,智商在29~79,90%的患儿常有言语发育落后,并且随着年龄的增长,可能出现认知及言语功能的倒退[7]。除了生长发育迟缓、智能低下等症状外,临床医生仍需要密切关注罕见病儿童的精神行为发育和心理卫生健康,遗憾的是目前关于此类研究的相关报道较少。RSTS患儿的精神行为异常发病率也显著高于正常同龄儿童,如孤独症谱系障碍、多动症、注意力缺陷、强迫症等[7-12]。超过90%的RSTS患儿可以生存到成年,但约32%的患者生活能力日益减退,约37%的患者行为功能缺失愈加严重[13]。临床治疗宜早期干预以期延缓患儿生活能力的减退,并且对患儿的精神状态亦不能忽视。
RSTS系常染色体显性遗传性疾病,与2个致病基因突变相关:编码cAMP调节的增强子结合蛋白的CREBBP基因和编码E1A结合蛋白(p300)的EP300基因。研究发现CREBBP突变约占60%,为RSTS-1;EP300突变约占10%,为RSTS-2,仍有30%的病例未找到明确致病基因[14-17],目前未见种族及性别差异的相关报道。CREBBP基因位于染色体16p13.3区域,长度为156 kb,包含2 442个氨基酸的增强子结合蛋白;EP300基因位于染色体22q13.2,长度为88 kb,包含2 414个氨基酸的p300蛋白,两者由相同数量的外显子(31)组成。增强子结合蛋白和p300蛋白包含了组蛋白乙酰转移酶活性区、非组蛋白乙酰转移酶活性区及多个蛋白结合区,二者均参与RNA聚合酶Ⅱ复合物和DNA结合转录因子的生物反应,在p53的泛素化和降解过程中发挥重要作用,对DNA修复、生长和分化、细胞的凋亡以及肿瘤因子的抑制[18]等方面有积极的调节作用。CREBBP和EP300均编码组蛋白乙酰转移酶,引起组蛋白末端赖氨酸的电荷性质发生改变,减弱了DNA-组蛋白的相互作用,促进遗传物质转运[19]。CREBBP及EP300是一对高度同源化的基因,有70%以上的序列同源性,二者的反式激活结构域不与特定的DNA结合,参与编码高度同源性的蛋白,与人体的转录因子及基因蛋白编码区相互影响。既往研究认为剂量效应的变化可以引起基因组拷贝数发生变异从而造成发病,而CREBBP基因作为一种剂量敏感基因,编码蛋白在目的DNA与转录因子之间发挥着枢纽作用。如果CREBBP基因位点发生变异,会改变蛋白的结构而致病,并且片段缺失也会引起单倍剂量的减少,从而影响蛋白的功能而引起正常生理功能发生变化[20]。CREBBP基因的突变形式有230余种,包括无义/错义突变、剪接位点突变、缺失突变、异位重排、插入、大片段插入/重复突变等。EP300基因相的突变形式有9种,包括无义/错义突变、缺失突变[21]。基因突变类型与临床表现的相关性目前没有定论,未见CREBBP基因、EP300基因突变导致不同临床特征的相关报道。有研究认为CREBBP基因缺失片段的大小与病情的严重程度相关,但也有学者指出认为CREBBP基因各类突变导致的畸形面容并无明显差别[22-23]。张江伟等[21]报道过2例RSTS患儿,缺失突变引起的症状较轻,而错义突变引起的症状相对较轻,这与既往研究观点不同。本病的预后尚缺乏大样本的长期随访调查,相关报道较少,仅有研究表明超过90%的患儿可以活到成年期。智力障碍、癫痫、情绪障碍、孤独症行为等将长期存在,大部分患儿无法获得独立生活的能力,无法上学和社交,预后较差。RSTS作为一种罕见病,早期诊断,尽早治疗能积极改善患儿的预后,但由于缺乏明确的诊断标准,目前临床上主要依据症状特征及基因检测技术来进行诊断。近年来全外显子组测序检测技术在罕见病的诊断应用方面取得了积极的进展。全外显子测序技术对目标序列的覆盖度约为99%,目前可以检测人类近2万个有功能的基因的外显子编码区序列。对于不明原因发育迟缓或智力障碍的儿童,同时伴有宽大的拇指和(或)脚趾,或具有特殊面容及其他脏器畸形,应积极进行全外显子测序检测,以提高RSTS的诊断率,这对于其他神经系统罕见病的早期诊断亦大有裨益。目前RSTS的诊断水平及发病机制相关研究取得了一定成果,但仍有诸多问题亟待研究,如继续拓展RSTS的基因表型,深入识别RSTS基因型与表型的相关性,通过全外显子测序等高通量测序技术探寻除CREBBP和EP300外的新的致病基因。只有不断提高RSTS的诊断及机制研究水平,才能持续改进该病的治疗方案,促进治疗效果,改善疾病预后。此外,还可以为家庭再次生育提供遗传咨询和产前诊断,减少RSTS发病率。
