改性TiO2固体酸的制备及酸催化性能研究进展
2021-08-30周硕林游高林刘贤响尹笃林
周硕林,游高林,刘贤响,尹笃林
(1. 长沙师范学院 基础实验中心,湖南 长沙 410100;2. 湖南师范大学 化学化工学院 石化新材料与资源精细利用国家地方联合工程实验室,湖南 长沙 410081)
传统液体酸催化剂在反应体系中能与反应物充分接触、酸性强、活性高,但存在对设备腐蚀作用大、反应后催化剂与反应体系分离困难、易对环境造成污染等问题。随着绿色催化概念的提出,固体催化剂取代液体催化剂成为大势所趋。TiO2作为固体酸催化剂受到了国内外研究者的广泛关注,但TiO2在应用中仍存在一些局限性。
本文介绍了TiO2固体酸的局限性,综述了改性TiO2固体酸的制备方法及其酸催化应用的研究进展,分析了这些制备方法的优缺点及存在的问题,并在此基础上对今后的研究方向进行了展望。
1 TiO2固体酸的局限性
TiO2固体酸表面的L酸位点是酸催化活性的关键,并且在水中具有较好的稳定性。TiO2固体酸因价格廉价、催化剂与反应体系易分离等优点而越来越多地应用于有机合成中。但TiO2固体酸还存在以下局限性:1)表面的酸活性中心数目有限,且酸强度不高,在一些催化反应中存在用量高,甚至催化活性不高等问题。2)表面的酸活性中心种类单一,为典型的L酸催化剂,尽管有文献表征出有B酸位点存在,但仍以L酸为主体,因此在需要B酸或B酸和L酸位点共存的催化反应中适应性不强。3)表面具有亲水性,在有机催化反应体系中分散性差,易自身团聚,导致与反应底物接触不理想,在一定程度上影响催化性能。
2 改性TiO2固体酸的制备及其酸催化性能
2.1 无机酸改性TiO2固体酸
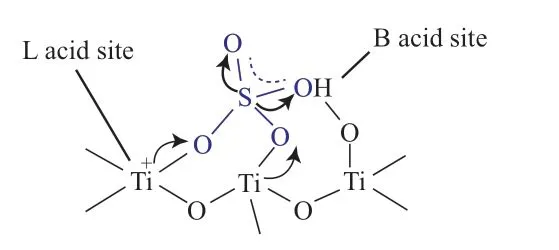
图1 SO/TiO2中酸活性位点结构[4]Fig.1 Illustration of the B and L acid sites in SO/TiO2 catalyst[4].
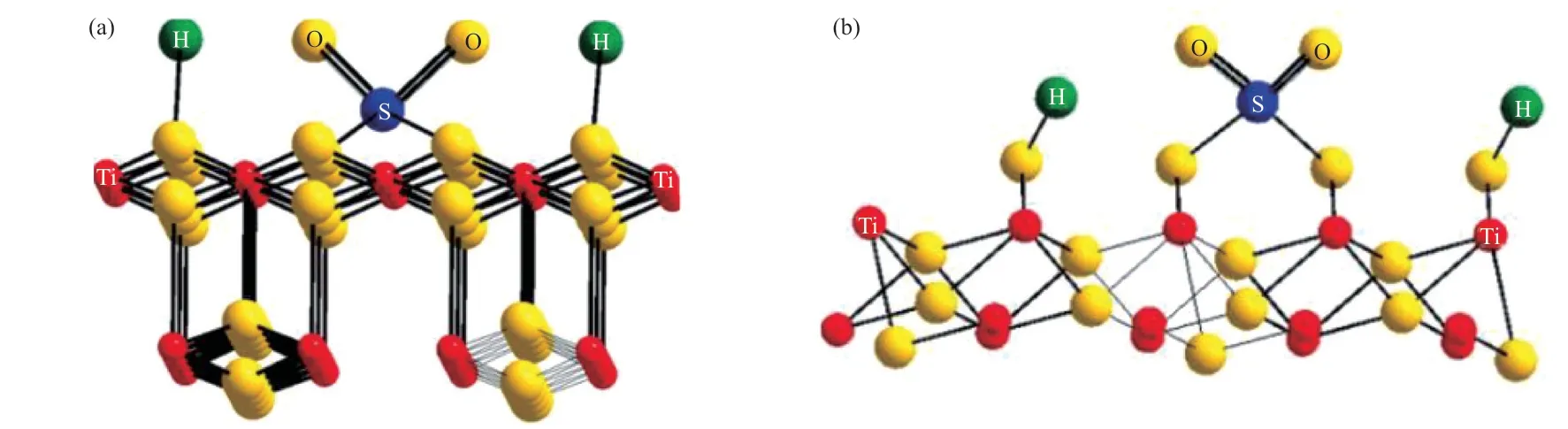
图2 SO/TiO2催化剂上硫酸盐的结构[5]Fig.2 Surface structures for the sulfate formation[5].
TiO2晶型及形貌对酸活性的影响也很显著。Zhao等[14]对比了多种形貌(钛纳米棒、钛纳米管、钛纳米粒子)和不同晶型(锐钛矿型、金红石型)的影响,发现表面酸位点的浓度是影响催化活性的主要因素,不同形貌TiO2的的催化活性的高低顺序为:纳米粒子>>钛纳米管>钛纳米棒,且单位面积的表面酸位点浓度与金红石型TiO2的含量呈线性正相关。而Nagarajan等[15]研究发现,相比钛纳米粒子,钛纳米管制备的催化芳香族或脂肪族羧酸与芳香胺反应的活性最高,酰胺化产物收率达75%~95%。Shao等[16]对比了不同浓度硫酸浸渍得到的硫酸钛纳米片催化剂的酸性特征。实验结果表明,随着硫酸浓度的升高,催化剂酸位点数量先增加后减小,并且以形成强酸为主。采用1 mol/L的硫酸浸渍时,催化剂酸量最高达139.7 μmol/g,此时表面B酸与L酸的比值最大为18.5,说明硫酸处理后主要形成B酸中心,这为进一步精细调控催化中心的类型、比例和酸强度提供了依据。硫酸钛纳米片催化醇解糠醇生成乙酰丙酸乙酯的收率达84.2%,且催化果糖转化成5-羟甲基糠醛(HMF)的收率达79%。
构筑结构新颖的TiO2可能会增加硫酸基团的数量或形成特殊结构的酸活性中心,从而提高酸催化性能。于荟等[17]报道了以无定形、高比表面积的水合钛酸为前体,采用等体积浸渍法制备新型晶须状介孔固体酸,该固体酸的酸催化活性远高于在最优条件下催化正丁醇和乙酸的酯化反应中,正丁醇转化率达94%,乙酸正丁酯选择性为100%。Li等[18]以自制钛酸钾为前体得到新型介孔TiO2(B)晶须,再通过硫酸浸渍法制备了高活性固体超强酸锐钛矿型TiO2的1.8倍,且B酸与L酸的比值为2.4,高于锐钛矿型TiO2中B酸与L酸的比值。与锐钛矿型TiO2相比,以为催化剂的乙酸与正丁醇的酯化反应速率更高,苯甲醚与苯甲醇烷基化反应时副产物更少。作者认为新型介孔TiO2(B)中较多的Ti与硫酸根配位形成独特的酸活性位点是影响催化性能的关键因素[18]。固体酸在催化酯化、转酯化等多类有机合成反应中表现出良好的应用前景[19-23],如表1所示。
表1 SO/TiO2在酸催化反应中的部分应用Table 1 Applications of SO/TiO2 in acid-catalyzed reactions
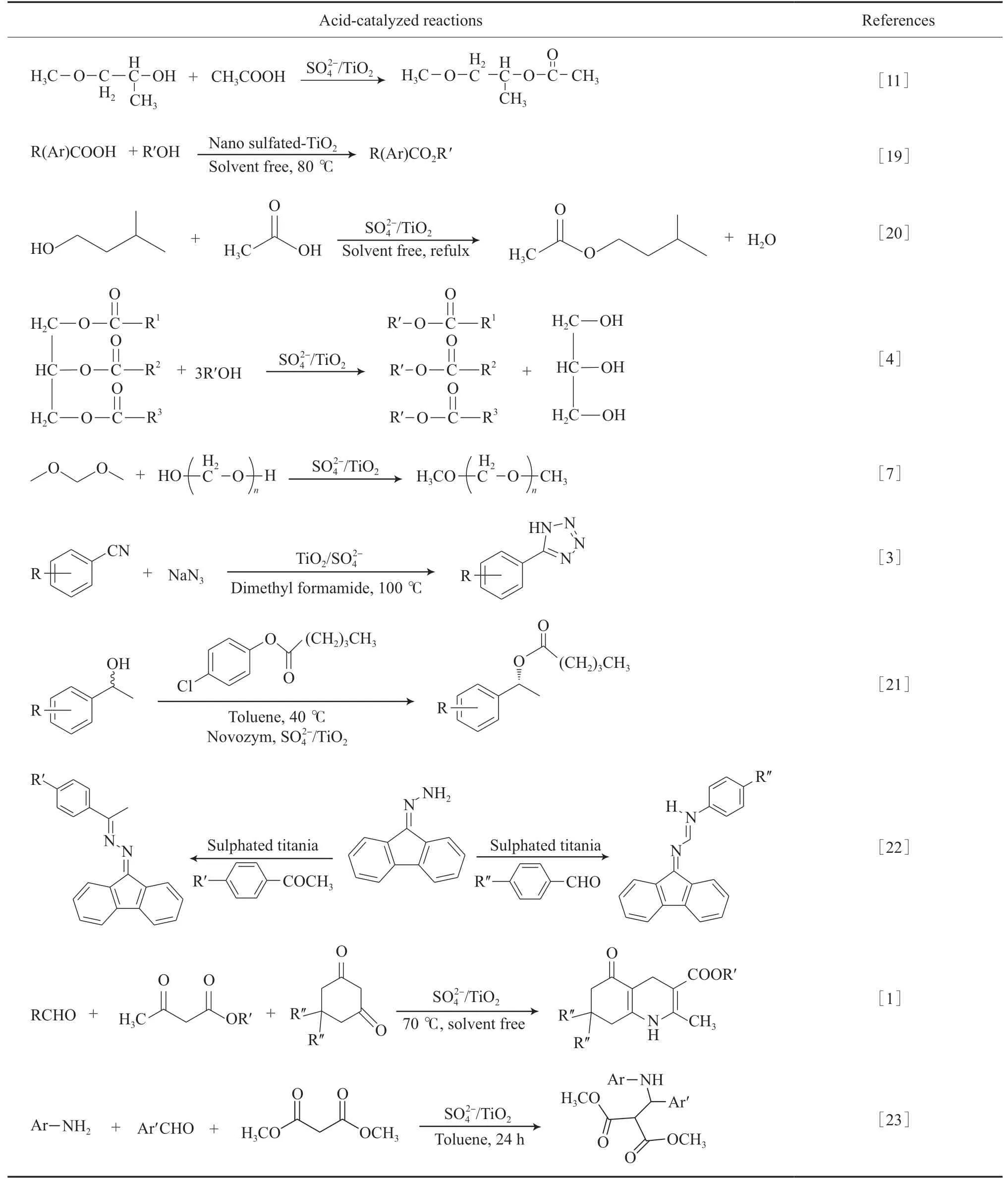
表1 SO/TiO2在酸催化反应中的部分应用Table 1 Applications of SO/TiO2 in acid-catalyzed reactions
Acid-catalyzed reactionsReferences HC OH+CH3COOH SO42-/TiO2 O C H2 HC O O H3CH3C H2 O C C CH3[11]CH3CH3 R(Ar)COOH + R'OH Nano sulfated-TiO2[19]Solvent free, 80 ℃R(Ar)CO2R'O O+SO42-/TiO2 Solvent free, refulx+[20]HOH3CH3CH2OO OH O H2C O C R1 O HC O C O H2C O C R2 R3+3R'OH O R'O C R1 H2C OH O SO42-/TiO2[4]O C R'+HC OH O R'O C R2 R3 H2C OH OO+C HO H2 H2CH3 H3CO O H SO42-/TiO2[7]C n O n R CN+NaN3 HN N NN TiO2/SO42-[3]Dimethyl formamide, 100 ℃R R OH O (CH2)3CH3 Cl O Toluene, 40 ℃Novozym, SO42-/TiO2 R O O(CH2)3CH3[21]R''R'N NH2 H N N N Sulphated titaniaSulphated titania N R'R''COCH3 CHO[22]RCHOH3C+O O O O COOR'SO42-/TiO2[1]R''OR'R''+R''O 70 ℃, solvent free R''NHCH3 O O Ar NH SO42-/TiO2 H3CO Ar'NH2 ArAr'CHO ++H3COOCH3[23]Toluene, 24 h O OCH3 O

磷酸或磷酸盐也可用来改性TiO2粒子。Dutta等[43]以钛酸异丁酯、磷酸为无机源,P123为模板剂,通过慢蒸发法制得磷酸钛。微波辅助下磷酸钛在二甲基乙酰胺-LiCl混合体系中能在较短的反应时间内有效催化果糖、葡萄糖等转化成HMF。高比表面积和表面数量可观的L酸位点是磷酸钛酸催化活性较高的原因。Atanda等[44]利用凝胶-溶胶法制得TiO2,再用磷酸铵水溶液浸渍、干燥后制得磷酸钛。相比TiO2,磷酸钛中L酸位点数量减少,而B酸位点数量增加,由此可见,TiO2经磷酸盐改性后可调变表面酸活性中心类型,实现由L酸到B酸中心的转变。他们在含有N-甲基-2-吡咯烷酮的水-四氢呋喃两相反应体系中,验证了磷酸钛能有效催化各种糖类化合物转化成HMF。在175 ℃下催化果糖、葡萄糖转化成HMF的收率分别达98%和90%;在180 ℃下催化纤维二糖、蔗糖转化成HMF的收率分别达94%和98%。可以看出磷酸或磷酸盐改性TiO2粒子后,所形成的酸中心种类可能存在差异。
2.1.4 TiO2-SO3H
氯磺酸是一种反应能力极强的磺化试剂,在冰浴条件下TiO2粒子与氯磺酸反应,得到TiO2-SO3H,TiO2表面通过引入磺酸位提高了酸催化活性,如图3所示。
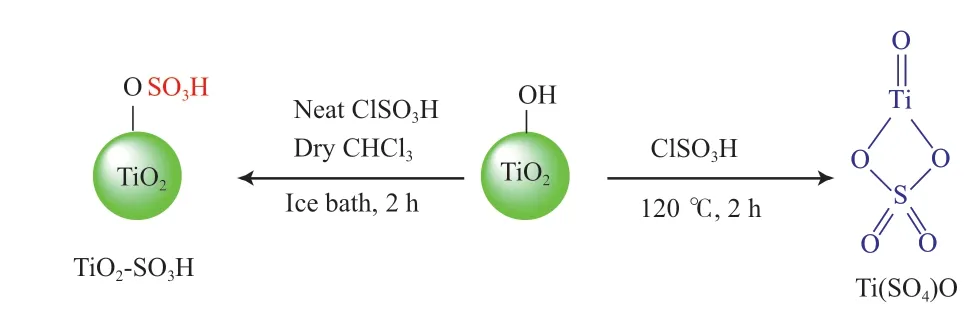
图3 TiO2-SO3H催化剂的制备Fig.3 Preparation of TiO2-SO3H catalyst.
Shirini等[45]报道了在水环境下,TiO2-SO3H在催化合成双香豆素衍生化合物中展现出良好的活性,催化转化频率达2 250 h-1,且可重复性较好,连续反应6次后,产物收率仍维持在95%左右,说明TiO2-SO3H在催化有机合成反应中是相对稳定的。Rahmani等[46]制备的TiO2-SO3H的酸强度为1.12,无溶剂条件下,在催化合成苄嘧啶酮、苯并噻唑、查耳酮衍生化合物时表现出优异的催化性能。用磁性Fe3O4粒子包覆TiO2后,再经氯磺酸修饰,可实现催化剂的磁性分离和回收[47]。Amoozadeh等[48]利用氯磺酸修饰TiO2得到的催化剂酸量为4.5 mmol/g,在无溶剂条件下该催化剂催化合成1,8-二氧-八氢氧杂蒽衍生物和四氢苯并[b]吡喃衍生物的收率分别为83%~96%,74%~98%。值得注意的是,在120 ℃下回流2 h,TiO2与氯磺酸反应可得到结构完全不同于TiO2粒子的硫代硫酸钛固体超强酸[49],物相组分为Ti(SO4)O,SO以化学键形式键合到TiO2晶相中,在甲醇与食用油摩尔比为9∶1、Ti(SO4)O用量为1.5%(w)、75 ℃、反应3 h的条件下,脂肪酸甲酯的收率高达97.1%。从图3的模型可看出,氯磺酸在不同条件下处理TiO2后形成的酸催化位点有较大差异。
2.1.5 TiO2-HClO4
高氯酸作为无机含氧强酸浸渍TiO2也可制备固体酸。Shirini等[50-51]用HClO4溶液浸渍TiO2,利用TiO2表面羟基引入酸催化位点,从而制得TiO2-HClO4(见图4)。该催化剂在醇或酚的化学选择性保护及去保护反应和选择性N-boc保护氨基反应中均表现出优异的催化性能。Shaterian等[52]利用TiO2-HClO4高效催化合成了1-酰氨基烷基-2-萘酚系列、1-氨基甲酸酯-烷基-2-萘酚系列、1-(a-氨基烷基)-2-萘酚系列和12-芳基-8,9,10,12-四氢苯并[α]-呫吨-11-酮系列化合物。TiO2-HClO4对不同底物均具有良好的催化性能,表现出普适性,且重复使用6次后仍有可观的催化活性。尽管TiO2-HClO4制备过程简单,不需高温焙烧,在有机合成反应中表现出优异的催化性能,但高氯酸是具有强腐蚀性和刺激性的一种危险化学品,寻求更安全、绿色的途径是构建酸活性位点的发展趋势。
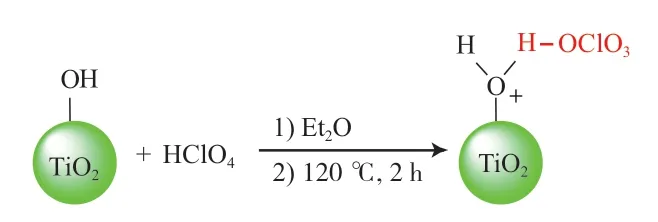
图4 TiO2-HClO4催化剂的制备[51]Fig.4 Preparation of TiO2-HClO4 catalyst[51].
2.2 有机-无机杂化改性TiO2固体酸
利用磺酸内酯化合物开环从而衍生出磺酸基团的方法在制备磺酸型离子液体中也得到广泛使用[53-55]。Gardy等[56]在甲苯回流下利用TiO2与1,3-丙磺酸内酯发生反应,可一步得到TiO2/PrSO3H固体酸催化剂(见图5),在TiO2表面键联强的有机磺酸基团为酸催化位。最优反应条件下,TiO2/PrSO3H催化已使用的食用油酯化和转酯化,脂肪酸甲酯的收率高达98.3%,且连续使用4次后催化活性没有明显下降,但在第5次使用时催化活性出现下降,他们认为可能与回收催化剂上的有机质或碳水化合物以及催化剂上丙基磺酸的流失有关。此外,1,4-丁磺酸内酯也是一种衍生磺酸官能团的前体。Hosseini等[57]在甲苯回流下用1,4-丁磺酸内酯与以钛酸四乙酯为前体合成的纳米TiO2反应,制得TiO2-Bu-SO3H催化剂,该催化剂的酸量达0.44 mmol/g。在无溶剂条件下,TiO2-Bu-SO3H在催化合成1-酰氨基-2-萘酚和1,8-二氧-八氢氧杂蒽衍生物中表现出良好的酸催化活性。
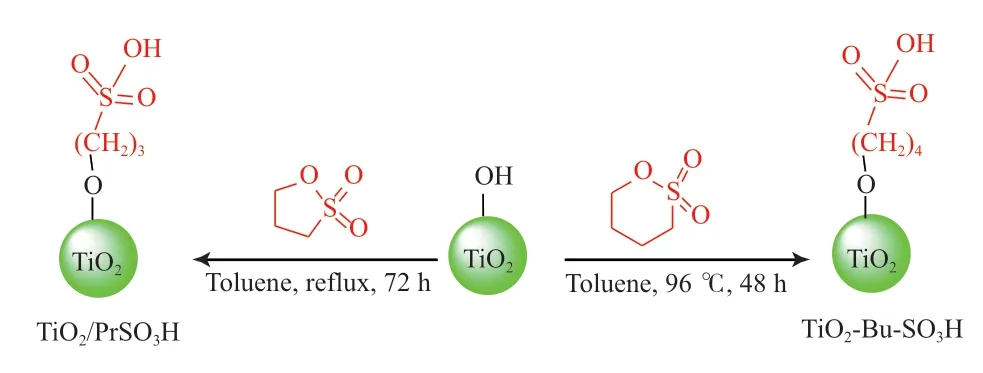
图5 TiO2/PrSO3H和TiO2-Bu-SO3H催化剂的制备[56]Fig.5 Preparation of TiO2/PrSO3H and nano-TiO2-Bu-SO3H catalyst[56].
在TiO2表面嫁接潜在的化合物后,再转化成酸性基团也是构筑酸位点的重要手段。其中,硫醇经氧化获得酸活性中心是构建酸催化剂的有效策略,可直接减少酸的用量,也可从分子水平上实现酸活性中心数量的调控。Atghia等[58]利用TiO2表面的羟基基团与3-巯基三甲基硅烷反应,然后在室温下用双氧水氧化,制得TiO2-Pr-SO3H催化剂(见图6)。该催化剂具有较好的热稳定性,酸强度为0.98,在较短的反应时间和较低的反应温度下能高收率地催化合成喹恶啉类化合物。同时,它还能快速催化合成香豆素类系列化合物,当用量为10 mg时,在较短的反应时间(22~90 min)下产物收率可达81%~95%。利用TEM和SEM对使用后的催化剂表征发现,催化剂使用4次后形貌未发生变化[59]。此外,该催化剂为叔丁氧羰基保护氨基提供了一条简单高效的路径,催化剂易从反应体系中分离回收,经丙酮洗涤、干燥后,重复使用次数可达15次[60]。Murugesan等[61]研究发现,在无溶剂条件下反应2 h,TiO2-Pr-SO3H可高收率合成哌嗪-喹啉基吡喃衍生物。TiO2-Pr-SO3H作为一种简单、安全、重复使用性好的固体酸催化剂,在有机反应特别在是多组分一步合成反应中展现出巨大发展潜力。

图6 TiO2-Pr-SO3H催化剂的制备[58]Fig.6 Preparation of nanoporous titania-based-sulfonic acid[58].
Shamsi等[62]利用纳米TiO2粒子与2,4-二异氰酸甲苯偶合后,将邻位异氰酸基团转化为氨基,再采用1,4-丁磺酸内酯经开环反应得到TiO2-NH-Bu-SO3H(见图7),表面引入的磺酸基团数量达0.6 mmol/g,可高收率催化合成四氢苯并[b]吡喃系列化合物,表现出较好的普适性。在此基础上,他们利用氨基官能团负载磷钨酸(HPW)构建酸活性位,制备了TiO2-NH2/HPW催化剂,该催化剂在35~65 min内合成2,4,5-三取代咪唑化合物的收率为85%~97%[63]。对TiO2表面进行修饰构筑酸催化位的策略,为构建多样化酸催化活性位点提供了有效途径,同时也为精细调控酸催化性能提供了思路。
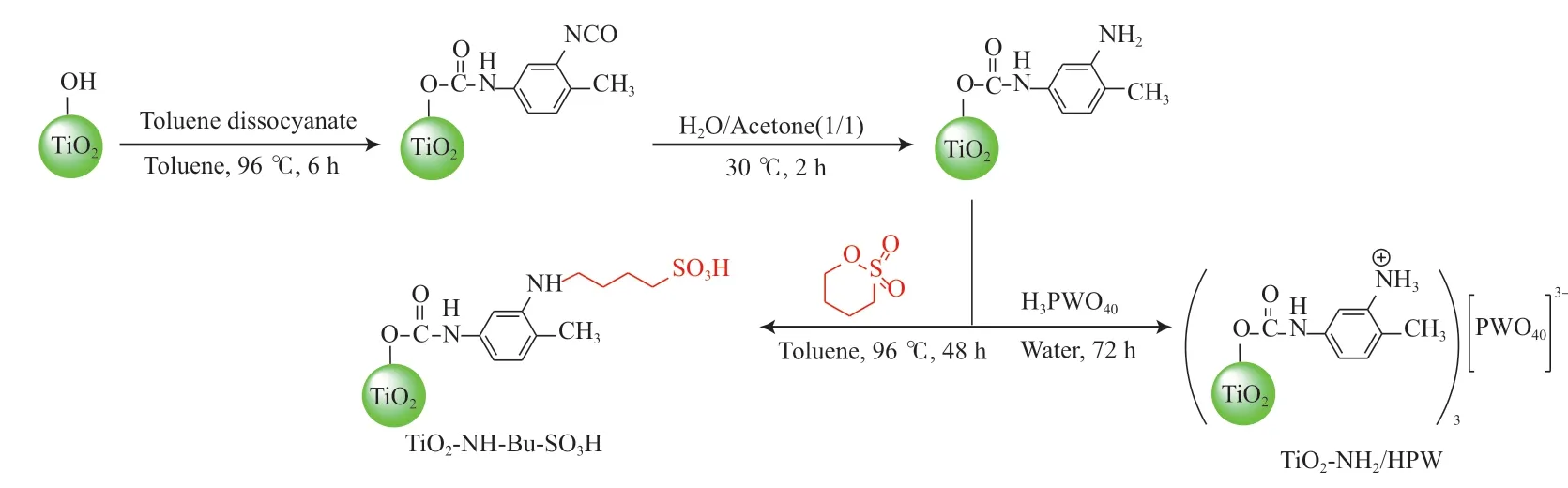
图7 TiO2-NH-Bu-SO3H和TiO2-NH2/HPW的制备[62]Fig.7 Preparation of TiO2-NH-Bu-SO3H and TiO2-NH2/HPW catalyst[62].
2.3 负载型改性TiO2固体酸
TiO2既是固体酸催化剂,又是优良的催化剂载体,在它的表面固载杂多酸催化剂可构筑复合型固体酸催化剂。周华锋等[64]等采用溶胶-凝胶法制得H4SiW12O40/介孔TiO2复合催化剂,在催化合成柠檬酸三丁酯反应中表现出良好的酸催化性能,重复使用6次后仍有较好的催化活性。TiO2载体固载杂多酸复合材料在提升酸催化活性和重复使用性等方面较单一材料的优势更明显。
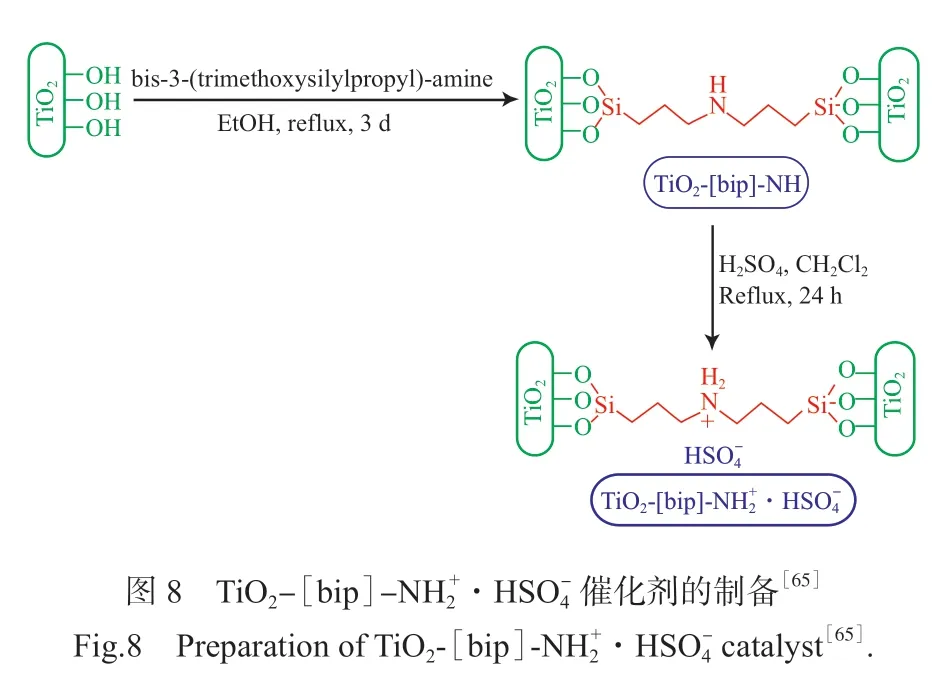
离子液体具有优异的柔韧性、耐热性、无腐蚀性和可调变性等,在TiO2上负载离子液体不仅可以实现离子液体的回收,而且还能在一定程度上实现催化性能的调控。Mazloumi等[65]在介孔TiO2上负载离子液体(见图8),所制备的催化剂在较短的反应时间、温和的反应条件下,能高收率合成N,N-二芳基甲脒衍生物、苯并䂳、苯并噻唑、苯并咪唑等化合物,并且通过过滤可从反应体系中回收催化剂,重复使用8次后,催化活性没有明显降低。离子液体在反应体系中不仅起催化作用,还起溶剂作用。根据催化体系调变离子液体的溶解性、设计离子液体的酸强度,正受到研究者的关注。
2.4 金属掺杂改性TiO2固体酸
金属氧化物或金属掺杂改性TiO2能通过调变表面酸活性中心种类、酸活性位点数量及强度调控酸催化活性。不同金属氧化物与TiO2结合时催化性能差异较大,其中,V2O5-TiO2[66]、WO3-TiO2[67]等二元金属氧化物是典型的固体酸。Madhuvilakku等[68]研究发现,ZnO-TiO2在棕榈油转酯化反应中表现出良好的催化活性,底物转化率最高98%,产物收率最高92%。Vilcocq等[69]研究发现,湿法浸渍制备的TiO2-WO3和共沉淀法制备的TiO2-ZrO2在酸性上有较大差异,焙烧温度对TiO2-WO3表面酸量的影响不大,但对TiO2-ZrO2表面酸量的影响显著,700 ℃焙烧下的TiO2-ZrO2表面酸量仅为600 ℃下焙烧的1/3。在TiO2-WO3催化纤维二糖水解的模型反应中,葡萄糖的选择性随掺杂剂自身性质和焙烧温度发生变化,葡萄糖收率可达60%。TiO2-WO3催化水解木聚糖的速度比催化水解纤维二糖快,水解产物主要为木糖和糠醛。Verma等[70]报道了铒掺杂TiO2粒子的固体酸,在多组分一锅合成多种螺环嘧啶吩嗪化合物中表现出优异的催化性能,这可能归因于Er3+取代部分Ti4+,形成了酸中心Ti—O—Er,但研究者未对酸中心种类、酸量及酸强度进行表征。Atanda等[44]比较不同化合物改性TiO2后表面酸中心类型及酸量的变化时发现,偏钨酸铵改性后,TiO2表面的L酸位点数量下降,B酸位点数量增加;偏钒酸铵改性后,L酸位点数量急剧下降,但B酸位点数量无明显增加;钼酸铵改性后,L酸消失,B酸位点数量显著增加。作者认为TiO2与氧化钼之间形成强的相互作用力是造成L酸转变至B酸的关键。Wada等[71]制备了铌掺杂的TiO2纳米管,发现掺杂少量铌后能增加B酸中心的数量,同时空心扭曲的纳米管结构有助于底物分子与酸中心结合,从而提升固体酸催化活性。
3 结语
对TiO2固体酸催化剂进行改性可进一步拓展固体酸的种类,提高固体酸在多种酸催化反应中的性能。其中,采用传统无机酸浸渍有利于酸位点的构建,但面临强腐蚀性、危险系数高等问题,同时在反应体系中催化剂酸活性位点存在不稳定性,寻求绿色、安全的途径仍是构筑酸活性位点的发展趋势。采用后合成策略制备有机-无机杂化TiO2固体酸催化剂,提供了多样化酸催化活性位点构建的途径,丰富了TiO2固体酸催化剂的种类。通过化学键合能有效实现载体与催化位点的稳定连接,能针对特定催化体系进行设计,并且通过优化合成条件进一步调控酸催化性能,极大拓宽了TiO2固体酸的催化应用范围。将离子液体、杂多酸、金属氧化物等与TiO2复合,由单一功能向多功能催化材料发展,有望提升催化剂的酸催化性能。此外,TiO2表面引入磁性材料对催化剂的分离和回收具有重要意义。由实心向介孔、空心等高比表面积的TiO2纳米材料发展,制备结构新颖的改性TiO2固体酸催化材料,有望获得优异的催化性能。借助先进表征手段,分析改性TiO2固体酸催化位点结构特征,研究酸位点构筑机制,提高酸催化位点稳定性,延长催化剂使用寿命仍是今后的研究重点。探索改性TiO2固体酸在绿色有机合成、生物质资源转化、耦合光催化协同催化等领域中的应用将是未来的热点方向。
