变异链球菌hit基因缺陷菌株的构建
2021-08-25赖扬帆王鹏乔里刘中静叶朝阳梁燕
赖扬帆,王鹏,乔里,刘中静,叶朝阳,梁燕
1.贵州医科大学附属口腔医院牙体牙髓科,贵州 贵阳(550004);2.贵州医科大学附属医院临床医学研究中心,贵州 贵阳(550004)
作为人类口腔中的主要致龋菌,变异链球菌大量繁殖形成牙菌斑生物膜附着在牙齿表面,其代谢形成局部酸环境长期作用于牙齿导致其脱矿,进而发生龋病[1]。目前对于变异链球菌基因功能研究仍主要集中在与其致龋性相关的生物膜形成、产酸耐酸“毒力因子”及其调控基因方面,如直接毒力侵染因子糖基转移酶(glucosyltransferases,GTFs)和葡聚糖结合蛋白(glucna-binding proteins,GBPs)。SMU.833糖基转移酶通过影响葡聚糖合成进而影响生物膜形成[2],SMU.940则调控葡聚糖结合蛋白C表达来影响生物膜形成[3-4]。变异链球菌胞壁(被膜)相关基因不仅可调控胞壁生物合成、细菌生长、分裂繁殖,也间接与致龋生物膜形成、产酸耐酸密切相关,如编码鼠李糖-葡萄糖胞壁多糖合成酶(rhamnose-glucose polysaccharide G,rgpG),编码生物膜调控蛋白(biofilm regulatory protein A,brpA)和编码青霉素结合蛋白5合成抑制蛋白(penicillin-sensitive repressor protein,psr)基因,当三者同时突变时,与标准株相比,三缺陷菌(rgpG/brpA/psr)不仅显著降低生长速率,还加剧菌体分裂“紊乱”形态(“短杆状”菌体“肿胀”变大,出现多重非对称中间隔),严重影响生物膜形成[5]。
变异链球菌基因组中,hit基因编码HIT家族蛋白SMU.412c,且该基因与brpA(lytR)基因邻近[6]。同时,生物信息学分析表明,HIT家族蛋白广泛存在于原核生物、真核生物、哺乳类动物,而且高度保守,具有重要生理功能[7]。hit基因是否可以调控变异链球菌增殖及生长周期尚不清楚。本研究利用基因同源重组方法敲除变异链球菌ATCC25175标准株hit基因片段,构建hit基因缺陷菌株,为后续研究变异链球菌hit基因与上述brpA、psr和rgpG等基因间互相作用,及其在变异链球菌生长繁殖以及生物膜形成、产酸耐酸等致龋性方面研究提供研究基础。
1 材料和方法
1.1 主要材料及试剂
BHI(brain heart infusion)培养基(杭州百思生物技术有限公司,中国);LB(Luria-Bertani)培养基(OXOID公司,英国);壮观霉素(上海生工生物工程股份有限公司,中国);十六烷基三甲基溴化铵(北京索莱宝科技有限公司,中国);限制性内切酶[宝生物技术(大连)有限公司,中国];质粒小提试剂盒(北京天根生化科技有限公司,中国);普通DNA产物纯化回收试剂盒(北京天根生化科技有限公司,中国);DNA maker(北京全式金生物技术有限公司,中国);厌氧产气袋(三菱集团,日本);质粒pFW5(上海沪震公司,中国);变异链球菌ATCC25175(NCTC10449)标准株(北京中科质检生物技术有限公司,中国)。
1.2 菌株及培养条件
变异链球菌ATCC25175(NCTC10449)标准株(类型:Streptococcus mutansClarke,来源:牙本质龋)培养条件:①培养基是BHI脑心浸出液肉汤,3.7 g/100 mL液体或1%琼脂粉培养基,按121℃/0.1 MPa灭菌20 min;②37℃,厌氧孵育,静置(或小摇床200 rpm)培养24~48 h。
大肠杆菌感受态DH5α(本实验室保存菌种)培养条件:①培养基:LB液体或1%(w/v)琼脂固体培养基;②37℃孵育静置(或小摇床220 rpm)培养过夜。
1.3 变异链球菌全基因组DNA提取
取变异链球菌ATCC25175甘油菌种,室温复苏,划线接种于BHI固体培养板,37℃厌氧培养48 h,挑取单克隆菌斑,接种于BHI液体培养基,24 h后取出,菌液离心后收集菌体,用CTAB法提取基因组DNA,-20℃保存备用。
1.4 基因扩增引物
变异链球菌ATCC25175标准株基因组全序:NCBI Reference Sequence:NZ_LS483349.1,大小为2 019 343 bp),其hit基因大小为420 bp;本课题研究hit基因上下游序列范围为:1664202~1665870,大小为1 669 bp。质粒载体pFW5序列:GenBank,ACCESSION:U41082,大小为2 726 bp[8],由上海沪震公司提供测序报告,利用生物软件Primer Premier 5.00设计相关PCR扩增引物序列,如表1所示。以提取的变异链球菌基因组DNA为模板,配制PCR反应扩增液,进行PCR反应,然后用1%(wt/vol)琼脂糖凝胶电泳鉴定。
1.5 重组质粒pFW5(hit-Up-pFW5-Down)构建
配制双酶切反应液,将hit基因上游片段与载体pFW5质粒进行双酶切,37℃酶切反应3~6 h,酶切产物使用普通DNA产物纯化试剂盒进行纯化,再分别检测pFW5质粒与hit上游片段双酶切后的浓度。
根据T4 DNA Ligase产品说明书,配置pFW5质粒与hit基因上游片段连接反应液。连接后的产物热激转入DH5α感受态细胞,涂布于Spe+(0.1 mg/mL)抗性LB固体培养基圆板,37℃倒置静置过夜培养,提取阳性单克隆菌斑质粒。同样的方法再将hit基因下游片段插入已构建好的质粒中,获得hit基因缺陷重组质粒(编号为20#和141#)。在pFW5载体的两个多克隆位点区(MCS-I和MCS-II)前后,设计了pFW5(PCR)两对引物(pFW5-F/pFW5-R1和pFW5-F1/pFW5-R,见表1),对20#、141#及对照空质粒pFW5进行PCR扩增双酶切鉴定,挑选出正确大小的质粒送检测序。测序结果在NCBI上进行Blast对比拼接。

表1 实验所用引物及其序列Table 1 Primers and sequences used in the experiment
1.6 变异链球菌hit基因缺陷菌株的构建
选取变异链球菌ATCC25175标准株单克隆菌斑接种入BHI液体培养基,培养至对数生长中期,抽取100μL菌液接种至2 mL BHI液体培养基,继续培养2 h,取300μL上述菌液加入2.4μL浓度为1 mg/mL的感受态刺激肽CSP溶液[9],轻轻混匀,孵育0.5 h。
将单酶切线性化纯化后的hit-Up-pFW5-Down质粒4μg加入到上述菌液中,轻轻摇晃培养3 h后将菌液涂布于Spe+(0.1 mg/mL)抗性的BHI培养板培养48 h。选取阳性单克隆菌斑,用CTAB法提取基因组,送检测序。
1.7 变异链球菌hit基因缺陷菌株遗传稳定性
将构建成功的变异链球菌hit基因缺陷菌株提高壮观霉素质量分数(1 mg/mL)后多次传代,提取基因组DNA,再次送检测序。
1.8 变异链球菌hit基因缺陷菌株生长曲线测定
①甘油菌种经BHI固体培养基划线活化后,挑选单克隆菌斑接种于BHI液体培养基,厌氧培养约20 h;取部分种子菌液,用无抗BHI培养基稀释到0.5麦氏比浊浓度,然后稀释100倍为工作菌液[(1~2)×106CFU/mL]。②取96孔细胞培养板,其中每孔加入180μL无抗BHI培养基和20μL工作菌液,厌氧培养,监测其在48 h内OD600值变化。设置三组平行实验,变异链球菌ATCC25175标准株进行生长对照。生长速率计算方法:生长速率=[(OD600)t-(OD600)0]/t。
1.9 统计学分析
采用SPSS19.0软件进行统计分析,定量资料服从正态分布,采用均数±标准差表示,组间比较采用独立样本t检验,P<0.05为差异有统计学意义。
2 结果
2.1 hit基因上下游片段扩增
以变异链球菌ATCC25175标准株基因组DNA为模板,分别利用hit上下游引物对,扩增hit基因上下游片段,hit上游片段大小为850 bp,下游片段大小为519 bp。扩增产物经核酸电泳鉴定,hit基因上下游扩增产物均为单一明亮条带,大小与预计大小相近(图1a)。质粒载体pFW5有上下两个多克隆位点(MCS-I和MCS-II),两者被壮观霉素抗性基因aad9所分隔开(图1b)。
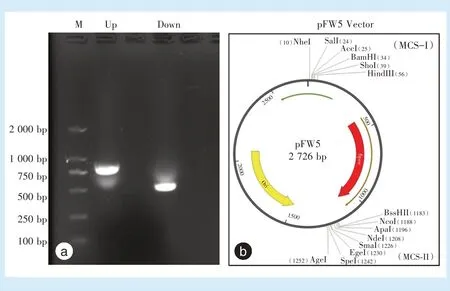
Figure 1 Electrophoresis of PCR products of the upstream and downstream sequences of the hit gene and the circle map of the pFW5 vector图1 hit基因上游、下游片段PCR产物电泳和pFW5载体图
2.2 pFW5重组质粒构建
重组质粒20#和141#PCR扩增电泳结果如图2a所示。在MCS-Ⅰ区插入hit基因上游片段部分PCR产物大小为1 774 bp;在MCS-Ⅱ区插入hit基因下游部分PCR产物为1 078 bp,其对应空载体pFW5为935 bp和571 bp,表明重组质粒上游部分插入约800 bp的hit基因上游片段和约500 bp的hit基因下游片段。
重组质粒20#和141#样品经XhoI和SpeI双酶切后电泳鉴定,未经酶切的空载体pFW5可见一条大小约2 700 bp明亮条带,与空载体大小相同;空载体pFW5经XhoI和SpeI双酶切后,两条带大小分别约为1 500 bp和1 100 bp,符合预期大小;重组质粒20#、141#经过双酶切后呈现两条清晰的条带,大小分别约为1 500 bp和2 500 bp,如图2b所示。
2.3 hit-Up-pFW5-Down重组质粒测序
为了精准确定重组质粒克隆构建,将pFW5(hit-Up-pFW5-Down)重组质粒20#、141#送公司测序,并将样本测序结果在NCBI数据库里进行BLAST(https://blast.ncbi.nlm.nih.gov/Blast.cgi)比 对和拼接。对比显示:上下游片段成功插入pFW5载体的MCS-I和MCS-II区间。变异链球菌hit基因缺陷重组质粒的连接顺序为:hit基因上游片段、Spe+抗性基因、hit基因下游片段,以上片段以正确序列与pFW5质粒连接构成基因缺陷质粒。
2.4 变异链球菌hit基因缺陷菌株筛选
将重组质粒转入变异链球菌后随机选取阳性单克隆菌斑若干,提取基因组DNA,进行PCR克隆鉴定,如图3所示。hit基因片段的PCR产物样本电泳图中,都出现大小一致明暗不同的两条带,分别约为1 400 bp和550 bp,提示pFW5重组质粒转入变异链球菌ATCC25175标准株后,部分与标准株本身hit基因实现同源重组;同时,也存在转入未被线性化重组质粒而又未实现同源重组菌株(菌斑显阳性,550 bp条带明亮),如20-8、20-9样本。
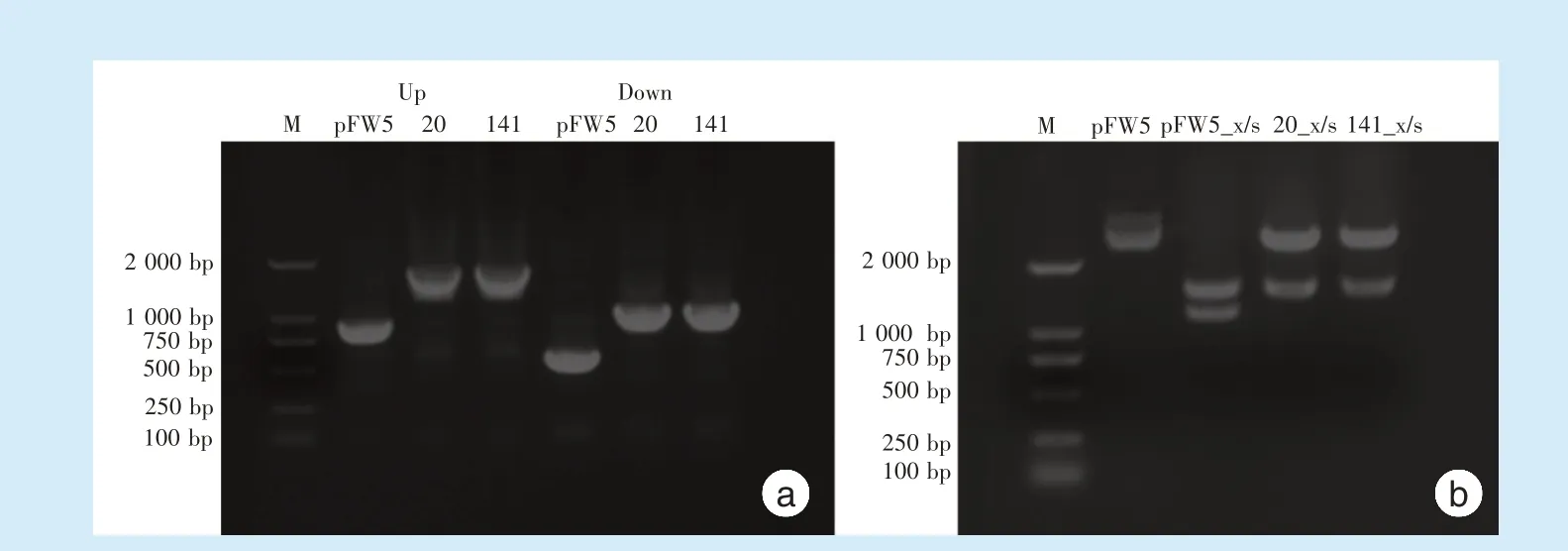
Figure 2 Electrophoresis of PCR-and double digested-products of the recombinant plasmid pFW5图2 重组质粒pFW5的PCR产物与其双酶切产物电泳
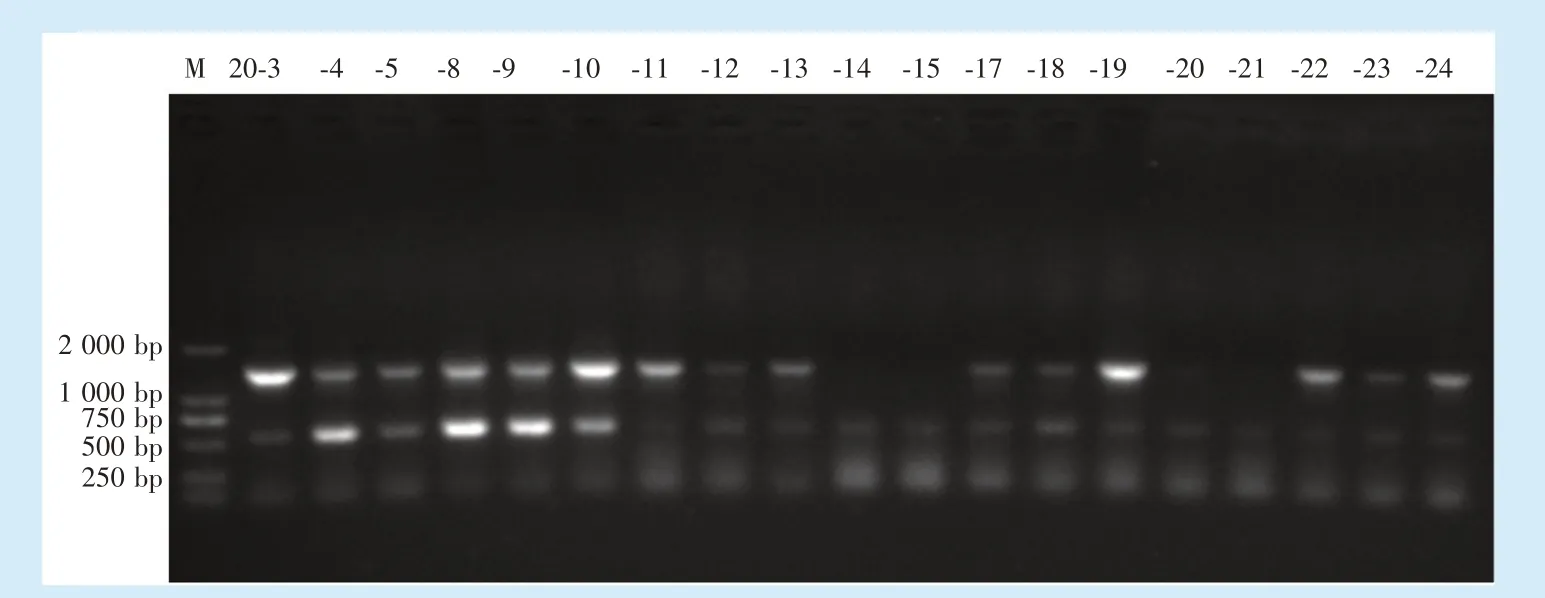
Figure 3 DNA electrophoresis of PCR products of the hit gene DNA fragments from the Streptococcus mutans genome of plasmid(20#)genetic transformation图3 质粒(20#)遗传转化变异链球菌株基因组的hit基因片段PCR产物核酸电泳
选择hit基因条带(550 bp)较弱或无该条带的样 品,如20-3#、20-5#等,并 对 照 变 异 链 球 菌ATCC25175标准株,进行hit基因片段PCR产物测序,其电泳条带和Sanger测序如图4所示。hit基因片段的PCR产物大小约为1 400 bp(图4a),而没有出现模式菌株ATCC25175大小约为550 bp的条带(图4c),表明原有的hit基因片段里都已插入了长达近900 bp碱基片段。PCR产物Sanger测序,正向测序读图显示:hit基因残余碱基序列片段碱基C(图4b,双箭头)后,紧接碱基序列片段的碱基A,该序列与载体pFW5上连接aad9壮观霉素抗性基因的连接序列相同,与相应位置处正常hit基因碱基序列明显对照(图4b,双箭头右侧部分)。
反向测序读图显示:hit基因残余碱基序列碱基C(图4d,双箭头)前,紧靠碱基序列碱基G,该序列与载体pFW5上连接aad9壮观霉素抗性基因的连接序列相同,也与对应位置处正常hit基因碱基序列明显对照(图4d,双箭头左侧部分),表明变异链球菌hit基因缺陷菌株构建成功。
2.5 变异链球菌hit基因缺陷菌株遗传稳定筛选
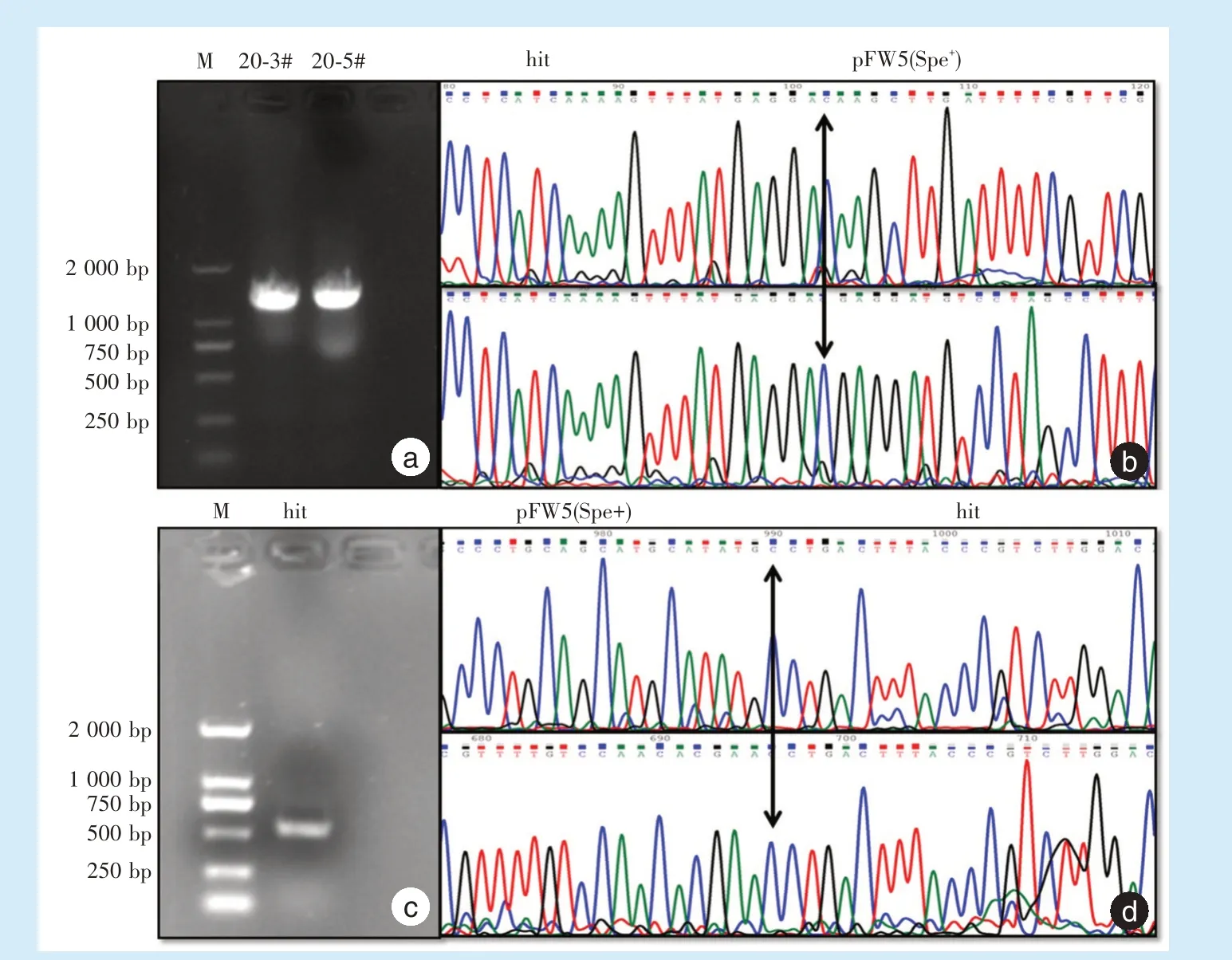
Figure 4 Electrophoresis and Sanger sequencing of PCR products of the hit gene fragment from the hit-deficient mutant strains and their parental S.mutans ATCC25175图4 变异链球菌hit基因缺陷突变菌株及其标准株ATCC25175的hit基因片段PCR产物电泳和Sanger测序图
将图3中的样品进行多次抗性筛选和鉴定,筛选后的缺陷菌株基因组的hit基因片段PCR产物电泳和Sanger测序,如图5所示。PCR产物电泳只有单一大小约1 400 bp条带(图5a)。PCR产物Sanger测序,正向测序显示:hit基因残余碱基序列碱基C(图5b,粗黑箭头)后,紧接碱基序列的碱基A,该序列为载体pFW5上连接aad9壮观霉素抗性基因的连接序列;反向测序显示:hit基因残余碱基序列碱基C(图5c,粗黑箭头)前,紧靠碱基序列碱基G,该序列也是载体pFW5上连接aad9壮观霉素抗性基因的连接序列,都与之前初筛时的Sanger测序结果相一致,表明获得遗传稳定hit基因缺陷变异链球突变菌株。
2.6 变异链球菌hit基因调控细菌生长周期
hit基因缺陷型变异链球突变菌株(Smu.Δhit)生长普遍都要比变异链球菌ATCC25175标准株更快速(图6):Smu.Δhit缺陷菌株迟缓期从15 h缩短到5 h左右;Smu.Δhit缺陷菌株进入对数期持续快速生长约12 h后,ATCC25175才开始从迟缓期进入对数生长期,持续快速生长约12 h后到达峰值,逐渐进入稳定期,此时生长速率开始下降,呈现比较典型细菌生长周期特征。而Smu.Δhit缺陷菌株在进入对数期持续快速生长约12 h后,生长速率出现较明显波动,约4 h后也达到其生长速率峰值,比较ATCC25175标准株提前约4 h进入生长速率下降的稳定期。从整个生长周期看来,Smu.Δhit缺陷菌株生长速率都要明显高于标准菌株Smu.25175(P<0.001),表明变异链球菌hit基因调控细菌生长周期。
3 讨论
本课题组利用基因同源重组方法,构建变异链球菌hit基因缺陷突变菌株,在BHI培养基中,hit基因缺陷变异链球菌株整个生长周期普遍比变异链球菌ATCC25175标准株更为快速,初步证实了GenBank注释所推测的调控细胞周期功能。
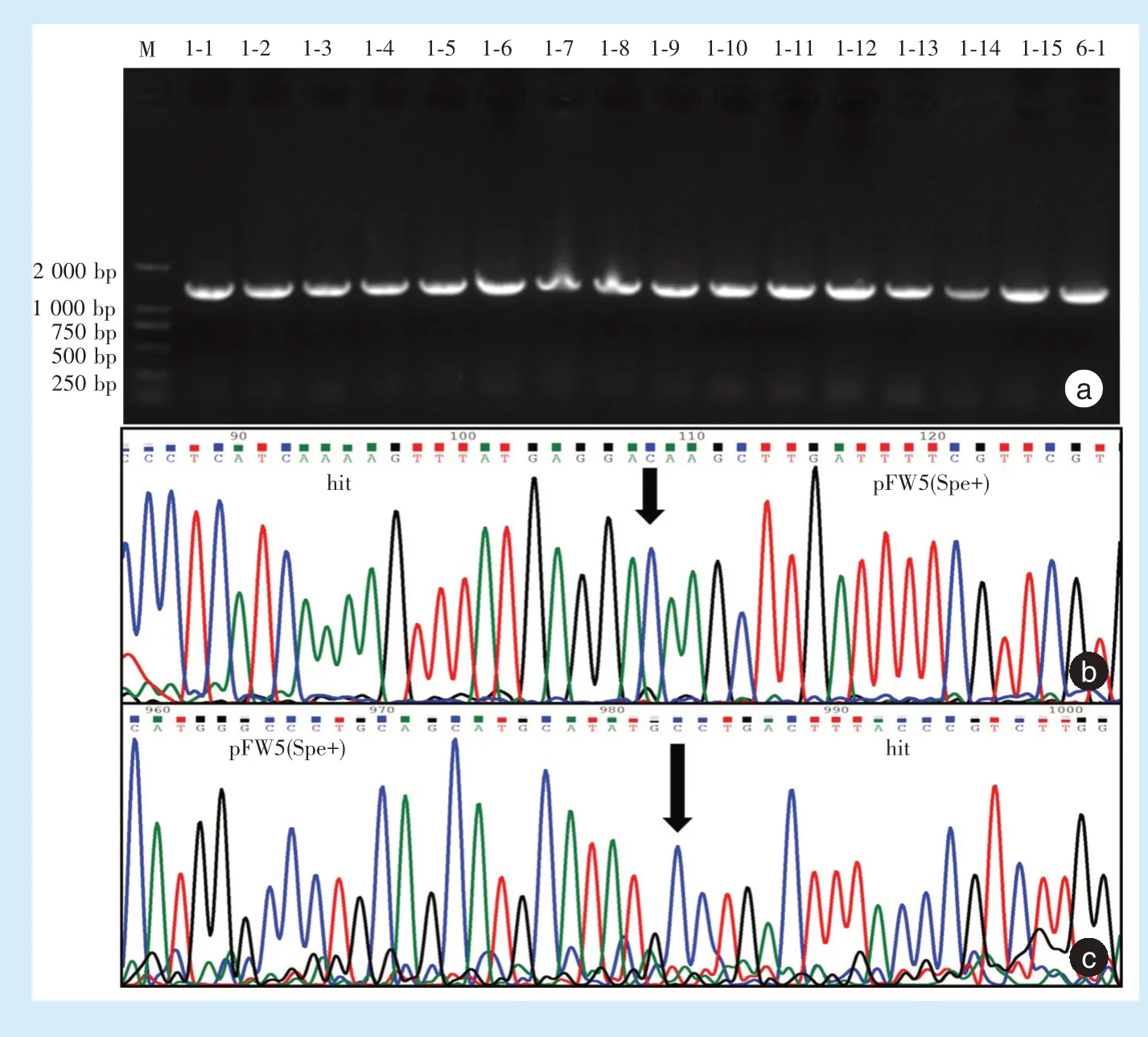
Figure 5 Electrophoresis and Sanger sequencing chromatograms of the PCR products of the hit gene DNA fragments from the hitdeficient mutant strains图5 遗传稳定hit基因缺陷变链突变菌株的hit基因片段PCR产物电泳和Sanger测序读图

Figure 6 Growth curve and growth rate of the hit-deficient mutant strains versus Streptococcus mutans图6 hit基因缺陷型变异链球突变菌株和标准株生长曲线和生长速率曲线
在ATCC25175标准株基因组全序中hit基因彼邻brgA(lytR)基因(通过调控自溶素酶表达来控制细菌生长周期[10])和ABC(ATP-Binding Cassette)相关基因,他们可能会组成一个“调控基因簇”,hit基因调控lytR(brgA)转录因子的转录活性。hit基因突变菌株成功构建,为hit基因在细菌生长繁殖致生物膜形成、产酸耐酸致龋性方面的研究提供条件。
细菌为适应环境变化,具有从环境中吸收和整合外源游离DNA的“天然遗传转化”机制,以获得新基因或新遗传性状,促进细菌抗抗生素和遗传变异的出现以及毒力因子快速进化[11-13]。这在链球菌中也广泛存在,如变异链球菌、唾液链球菌和肺炎链球菌中的“ComCDE基因簇”群体感应系统[14-15]。链球菌通过群体感应系统来诱导其进入遗传感受状态,主要是依赖于感受态刺激肽信号系统[16-17]。环境变化对其天然转化感受态产生巨大影响,细菌产生和分泌CSP信息素的能力也与转化效率相关,使用人工合成CSP可以避免细菌生产和分泌内源性CSP所需要的苛刻条件,人工合成CSP加入将转化效率提高了几个数量级,即使在野生型菌株中也是如此,扩大了可转化的菌株范围,在突变体构建中则可利用这一特性[18]。利用人工合成变异链球菌ATCC25175标准株刺激肽CSP(GenBank:SQF47995.1)成熟肽,诱导变异链球菌标准株进入遗传感受状态,利用该天然转化方法成功地使重组质粒转入标准株内进行同源重组,获得hit基因缺陷突变菌株。这与目前常用的电转化方法相比具有明显优势:无需专用电转仪,简单方便有效又不电击损伤变异链球菌体。本研究为深入了解变异链球菌hit基因功能及致龋性提供了基础。
【Author contributions】 Lai YF,Liang Y wrote the article.Lai YF,Wang P,Liu ZJ and Qiao L performed the experiments.Ye ZY designed the study,analyzed the data and revised the article.All authors read and approved the final manuscript as submitted.
