分子动力学模拟阴离子/两性离子表面活性剂在油-水界面的分子行为及协同效应
2021-08-24姁夏瑜苑莲花员润何绪文
吴 姁夏 瑜苑莲花员 润何绪文
(中国矿业大学(北京) 化学与环境工程学院,北京 100083)
表面活性剂因其独特的结构及其在溶液中存在的特殊形式和物理化学作用,应用范围日益广泛,主要用于农业[1]、纺织业[2-3]、石油工业[4-5]、医学[6]等领域。在石油工业中,表面活性剂作为采油的驱油剂,能较大地提高驱油效率,可使原油采收率提高[7]。然而在实际应用中,单一的表面活性剂几乎不能将油-水界面张力降低到预想的水平[8]。油-水界面表面张力的降低归因于界面和亲水基团之间的相互作用[9],而不同类型表面活性剂的混合体系通常表现出协同效应[10],因此,表面活性剂混合体系的性能研究近来受到广泛关注,许多研究集中在阴离子/非离子[11-13]表面活性剂混合体系和非离子/非离子[14]混合表面活性剂体系。Li等[15]研究了非离子表面活性剂辛基酚聚氧乙烯醚(Triton X-100)与阴离子表面活性剂十二烷基苯磺酸钠(SDBS)的混合体系,研究结果表明,在油-水界面中Triton X-100聚集在SDBS的空隙中,使得整个体系聚集更为紧密。Posocco等[14]研究了2种非离子表面活性剂吐温80(Tween 80)和司盘20(Span 20)的混合体系,研究结果表明,相对分子质量较小的Span 20会充斥于相对分子质量较大的Tween 80的缝隙中,这导致混合体系的界面排列比单一表面活性剂更为致密,2种表面活性剂分子之间具有显著的协同作用。Liu等[16]研究了不同碳链长度羧酸的混合体系与甜菜碱两性表面活性剂,研究显示混合效果主要受羧酸碳链长度的影响,甜菜碱表面活性剂分子的缝隙中充满羧酸分子,当加入适当长度的羧酸后,两者分子长度相协调导致分子间排列紧密,协同效应最佳。Zhao等[11]研究了十二烷基三甲基溴化铵(DTAB)、非离子表面活性剂烷基聚糖苷(APG)和十二烷基硫酸钠(SDS)的混合体系,研究结果显示,当3种表面活性剂按照一定比例混合时,3种分子可以交叉排列在界面处,在水相内交叉相聚形成胶束,混合体系性能更优越。但上述研究中没有具体研究哪种表面活性剂的亲水性/亲脂性更好。
目前,对阴离子/两性离子表面活性剂混合体系的研究尚少,与其他表面活性剂相比,阴离子表面活性剂具有更大的产量、更广的应用范围和更低的成本;两性离子表面活性剂具有水溶性好、刺激性低、抗硬水能力强等特点,可以弥补阴离子表面活性剂的不足,因此将阴离子和两性离子表面活性剂混合是非常必要的[17]。油-水界面张力的降低与界面性质有关,了解表面活性剂的结构及界面性质十分重要[18-19]。传统的实验方法不能解释混合表面活性剂之间的相互作用机理,随着计算机技术的快速发展,分子动力学模拟(MD)方法兴起,许多研究者将分子动力学方法用于研究油-水界面的微观作用机理和界面性质[20-24]。笔者选择石油开采中最常用的表面活性剂SDS/SB12-3[25-27]的混合体系,采用分子动力学模拟方法,研究了SDS与SB12-3在油-水界面的行为和协同作用机理。
1 表面活性剂在油-水界面性质的模拟方法
1.1 分子模型的构建
本次模拟使用Materials Studio程序,利用程序中的Visualizer模块分别搭建表面活性剂、正癸烷和水的分子模型。其次使用Forcite模块Geometric optimization(COMPASS力场[28-30])对各个单分子模型进行20000步的结构优化,使分子的能量最低,结构稳定。优化完成后SDS和SB12-3的分子结构如图1所示,为了便于观察,对氢原子进行隐藏,模拟结束后各原子所带电荷与其他文献相似[11,22]。
图1 动力学优化后表面活性剂分子结构Fig.1 Molecular structures of surfactants optimized by kinetics
根据常温常压下油和水的密度,利用AC模块构建了油相、水相,其中水相包含水分子2400个,水分子采用SPC模型[31],油相包含260个正癸烷分子,64个表面活性剂沿Z轴定向排列,最后通过Visualizer模块的Build layer工具将油相、表面活性剂和水相整合到1个盒子中,构成油-表面活性剂-水-表面活性剂-油的双界面模型,体系初始结构尺寸为5 nm×5 nm×12 nm(分别为X、Y、Z轴)。图2为初始SDS/SB12-3体系结构,初始体系表面活性剂尾部接近油相,亲水基团接近水相,可以使体系达到平衡的时间缩短。为保持体系的电中性,向水溶液中加入128个Na+和若干Cl-,各个模拟体系中2种表面活性剂的摩尔比及各个分子及离子的数目如表1所示。
图2 初始SDS/SB12-3体系结构Fig.2 Snapshots of the SDS/SB12-3 composite system
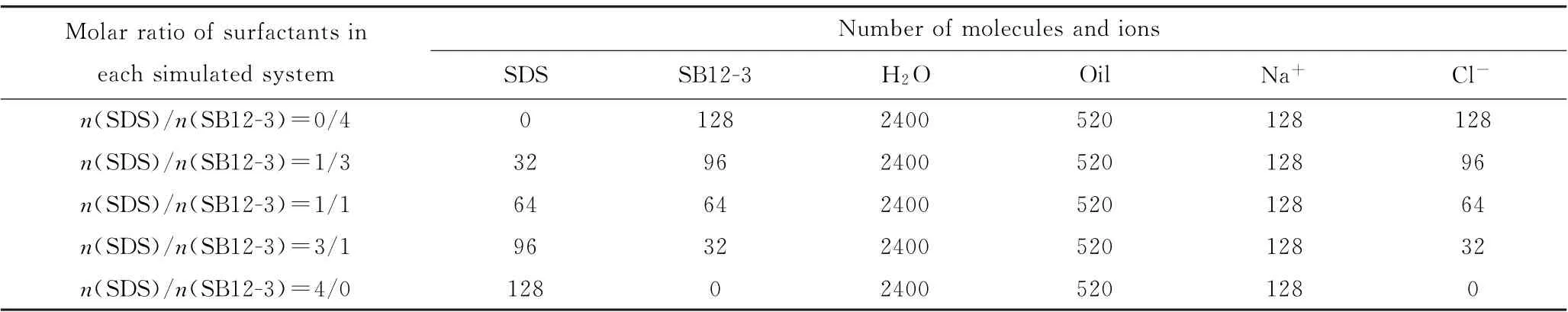
表1 表面活性剂在油-水界面模拟中各SDS/SB12-3混合体系的分子及离子数目Table 1 The number of molecules and ions in each SDS/SB12-3 composite system of surfactants in oil-water interface simulation
1.2 表面活性剂在油-水界面中性质的模拟细节
体系模型搭建好后,利用Forcite模块COMPASSII力场完成体系结构能量优化,采用Smart geometry optimization方法对各模型的初始结构进行优化,使能量达到最小化。Smart geometry optimization依次采用最陡下降法、共轭梯度法及牛顿法对体系进行优化,这是使体系能量最终达到最小化的一种智能优化方法;然后在Forcite模块里面Dynamics对能量优化后的体系进行模拟计算,选择正则系综(NVT),温度298 K,使用Nose-Hoover[32]恒温器,衰减常数0.1 ps[33],分子初速度随机产生[34]。截止半径不能大于盒子最小尺寸的一半,截止半径过小,计算精度不够;截止半径过大,计算结果错误,所以此次模拟选择截止半径为1.25 nm。静电相互作用使用Ewald法计算[35],模拟时长为10 ns,之后进行等温等压系综(NPT)模拟,条件压力0.0001 GPa,选用Berendsen[36]恒压条件下进行计算,时长10 ns,达到体系平衡。
2 结果与讨论
2.1 密度分布及界面形态
为了更清晰地分析表面活性剂在油-水体系中各组分的分布情况,给出了各体系沿Z轴方向的密度分布,如图3所示。其中表面活性剂的密度为体系中2种表面活性剂的平均密度,模拟得到的水相密度在0.992~1.002 g/cm3之间,这与水真实密度非常接近(0.997 g/cm3[37]),说明模拟体系的模型构建合理,力场选取和参数设置合适,动力学模拟结果可以反映各个体系的界面现象和聚集行为。图4为模拟后表面活性剂在油-水界面的平衡结构。可以看出:表面活性剂头基处于水相中,表明亲水头基进入到水层发生水化作用,带正电的Na+分布在带负电的头基周围,说明由于静电相互作用,Na+与表面活性剂极性头基结合。
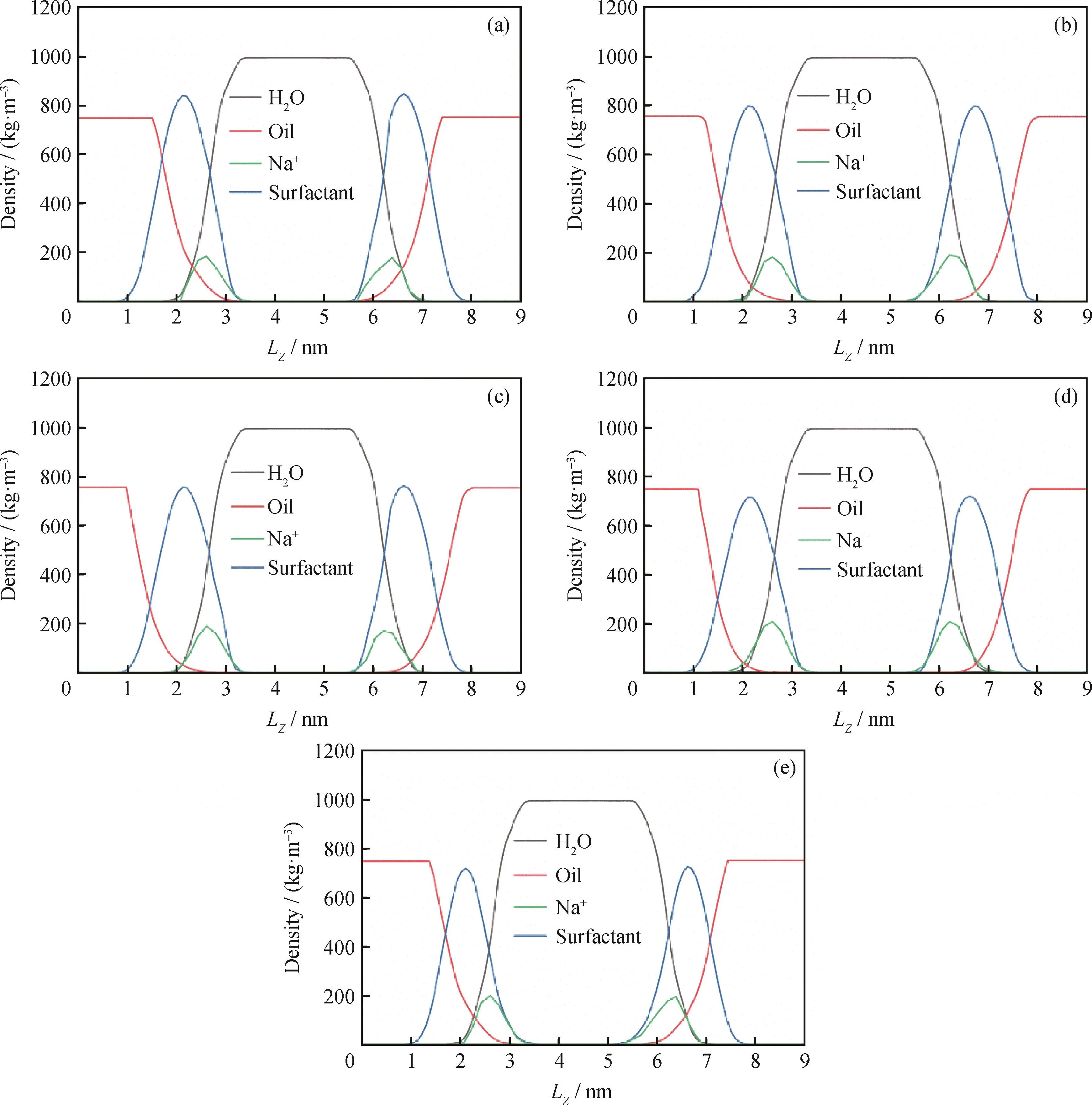
图3 不同摩尔比表面活性剂在油-水体系中的密度分布Fig.3 Density distributions of surfactants with different molar ratios in the oil-water system
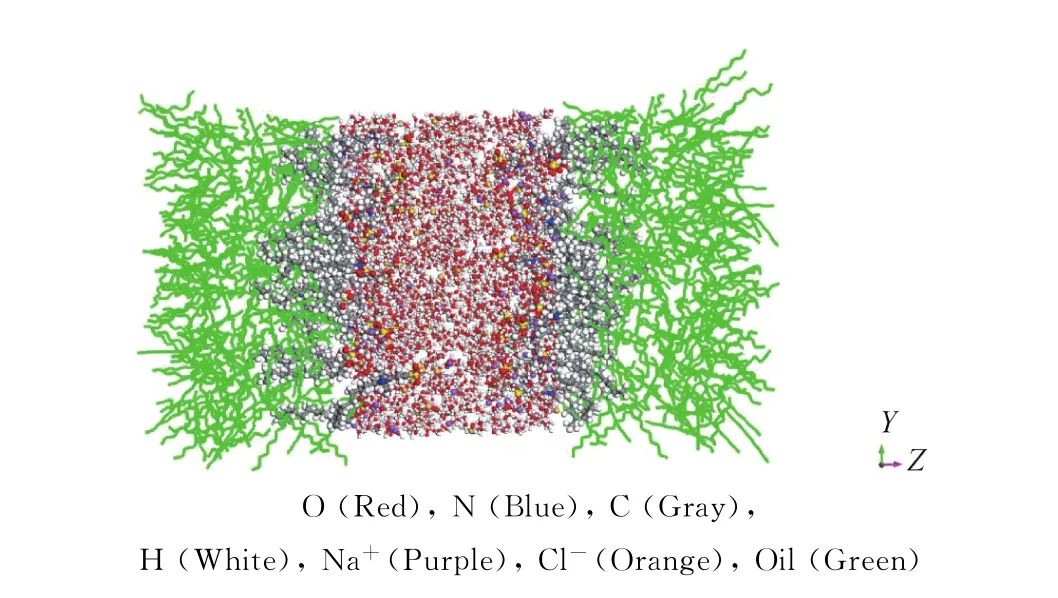
图4 模拟后表面活性剂在油-水体系中的平衡结构图Fig.4 The equilibrium structure snapshot of surfactants in the oil-water system after simulation
2.2 界面形成能和界面厚度
油-水界面表面活性剂的能量稳定性对界面性质有重要影响,其与表面活性剂分子之间、油与表面活性剂之间、表面活性剂与水分子之间的相互作用力大小密切相关,能够反映出界面体系的稳定性[38]。为了比较2种表面活性剂不同摩尔比体系的稳定性,计算了各个体系界面形成能(EIF),其计算公式如式(1)所示[39]。
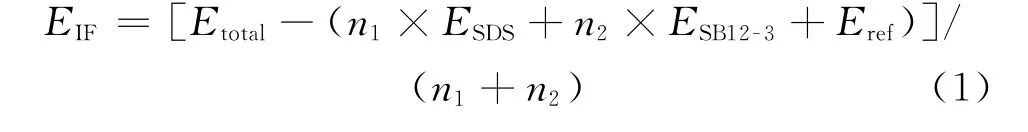
式(1)中:Etotal为表面活性剂在界面达到平衡后体系的势能,kJ/mol;Eref为无表面活性剂存在时只包括正癸烷-水界面体系的势能,kJ/mol;n1、n2分别为各个体系中SDS、SB12-3的数目;ESDS、ESB12-3分别为SDS、SB12-3的势能,kJ/mol,在模拟体系中仅在单个表面活性剂分子下计算。
表面活性剂的加入降低了界面张力,界面的总势能降低,因此EIF是负值,其绝对值越大,界面越稳定,对形成界面越有利[40]。表2为不同体系油-水界面厚度及界面形成能。可以看出,当n(SDS)/n(SB12-3)=1/1时,混合体系的界面形成能最低(-640.959 kJ/mol)。
界面厚度是界面的主要物理性质,可以直观地反映出界面吸附的强弱,是考察界面性质的一个重要指标,体系的界面厚度越大,界面活性越强[39]。对于油-表面活性剂-水体系的界面,通常采用“90%~90%”原则对界面厚度进行计算,即采用油相密度的90%到水相密度90%之间的距离定义为界面厚度[41]。由表2可见:n(SDS)/n(SB12-3)=1/1时,油-水界面厚度最大(1.942 nm),可能是因为SDS的—OSO3-和SB12-3上的—N+相互吸引,导致SDS和SB12-3不在同一表面上,使得2种表面活性剂在空间中的相对位置较远,界面粗糙度最大,界面厚度最大。混合体系中SDS过多时,虽然体系间还存在静电作用,但SDS比SB12-3分子长度短,造成界面厚度相对较小;若SB12-3过多的话,—N+占多数,静电斥力占主导地位,分子均匀排列,也会导致界面厚度稍小。5种表面活性剂混合体系的界面厚度由大到小的顺序为n(SDS)/n(SB12-3)=1/1、n(SDS)/n(SB12-3)=3/1、n(SDS)/n(SB12-3)=1/3、n(SDS)/n(SB12-3)=4/0、n(SDS)/n(SB12-3)=0/4,表明n(SDS)/n(SB12-3)=1/1时混合体系界面的稳定性高于单一体系和其他混合体系,界面形成能的变化趋势与界面厚度的变化趋势相一致。

表2 不同体系油-水界面厚度及界面形成能(E IF)Table 2 Oil-water of interfacial thickness and interfaceformation energy(E IF)of different systems
2.3 疏水尾链序参数
疏水基团和油分子之间的相互作用可以通过表面活性剂分子的头基和尾链之间的角度来分析,并且该角度还可以反映表面活性剂在油-水界面处的伸展性。图5为n(SDS)/n(SB12-3)=1/1时表面活性剂在油-水界面的平衡构型放大图。由图5发现,—OSO3-通过静电相互作用被吸引到SB12-3上的—N+附近,2种表面活性剂的尾部相互缠绕在一起,形成了紧密平行排列的结构,与上述界面厚度分析结果相一致。
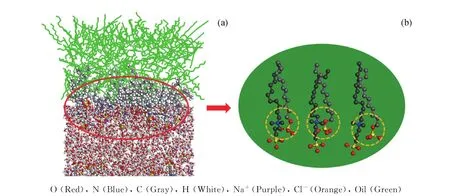
图5 表面活性剂在油-水界面的平衡构型放大图Fig.5 The enlarged equilibrium configuration snapshot of surfactants in the oil-water interface
图6为SDS和SB12-3的头基与尾链之间的夹角。可以看出,SDS和SB12-3的夹角分别为114.368°和111.903°,夹角越大,意味着烃尾链与油相的相互作用越强。SDS的角度大于SB12-3,说明SDS比SB12-3具有更好的亲脂性。表面活性剂的烃链的疏水尾链序参数(SCD)还可以表征疏水链的有序性,其计算公式如式(2)所示[42]。
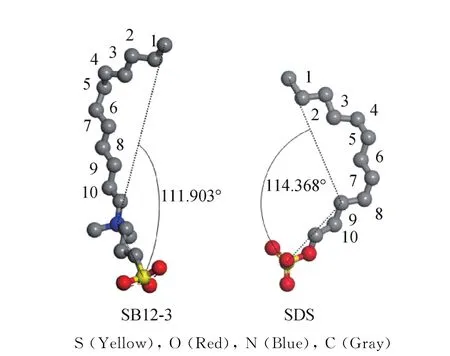
图6 SDS和SB12-3的头基与尾链之间的夹角Fig.6 Angles between the head base and the tail chain of SDS and SB12-3
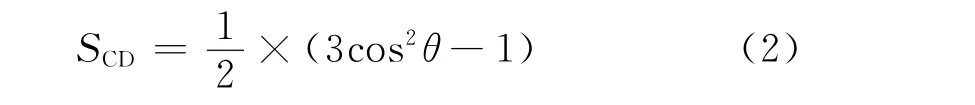
式(2)中:θ为Cn-1和Cn+1原子之间与界面法线(Z轴)的夹角。当SCD的极限为1时,表明烃链沿着界面法线方向(Z轴)完全有序;当SCD的极限值为-1/2时,尾链沿界面方向排列完美。SCD越大,说明尾链与界面法线的夹角越小且与界面越垂直;SCD越小,说明尾链与界面法线的夹角越接近90°,尾链与界面方向平行。
图7为表面活性剂疏水尾链序参数随碳原子数的变化。由图7可以看出:SDS和SB12-3的SCD从C1到C4呈上升趋势,且SB12-3的高于SDS,可以推断,SB12-3尾部烃链的这些碳原子在油-水界面上接近界面法线方向;在C5~C10之间,SDS的SCD大于SB12-3,这意味着SDS疏水尾链的这些碳原子的构象在油-水界面处更接近界面法线方向,并且SDS更接近法线方向的碳原子数目超过SB12-3,说明SDS在油相被拉伸得更多,具有更好的亲脂性,这与头基和尾链夹角的分析结果相一致。
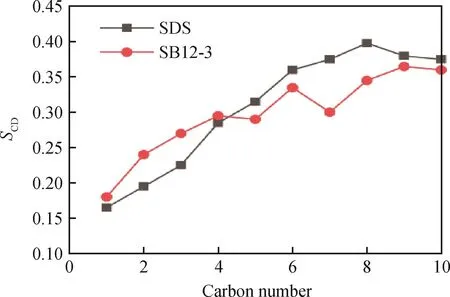
图7 表面活性剂疏水尾链序参数(S CD)随碳原子数的变化Fig.7 Deuterium order parameters(S CD)of the drain tail chain of surfactants vs carbon number
2.4 径向分布函数
在统计力学中,径向分布函数g(r)可以描述某种粒子的密度如何随着参考原子的距离(r)而呈现函数变化[43]。图8为H2O分子中的氧原子(OW)相对于混合体系的亲水基团(SDS中的—OSO3-和SB12-3中的—SO3-、—N+)中S和N的径向分布,分别用g1S-OW(r)、g2S-OW(r)和gN-OW(r)表示。由图8可见:g1S-OW(r)和g2S-OW(r)的第1个峰值出现在0.336 nm,第2个峰值出现在0.574 nm;g1S-OW(r)、g2S-OW(r)中第1个明显的峰表明2个头基与水分子间有很强的氢键相互作用,第2个弱峰分别由头基与第一水化膜分子之间的次强氢键相互作用所致;第1个峰与第2个峰的位置相差0.24 nm左右[44],恰好为1个氢键之间的距离,说明第1层水和第2层水之间也是以氢键的形式结合的。峰高可反映水分子与亲水性基团之间相互作用的强度,g1S-OW(r)的2个峰值分别为6.189和2.729,g2S-OW(r)的2个峰值分别为6.720和3.343;g1S-OW(r)的峰值均小于g2S-OW(r)的峰值,表明SDS与水分子之间的相互作用小于SB12-3与水之间的相互作用。gN-OW(r)第1个峰值出现在0.455 nm,比g1S-OW(r)和g2S-OW(r)出现峰值的r远,这一结果表明水分子优先与—SO3-和—OSO3-作用,并形成强大的氢键。—N+出现多个小峰,推测主要是由于—SO3-和—OSO3-的基团距离接近,水化膜在—N+周围产生部分重叠。为了进一步解释这一现象,笔者计算了氢键的数目,SB12-3与水之间的氢键数目(12.555)大于SDS与水之间的氢键数目(8.968),因此说明SB12-3在SDS/SB12-3混合体系中具有较好的亲水性。
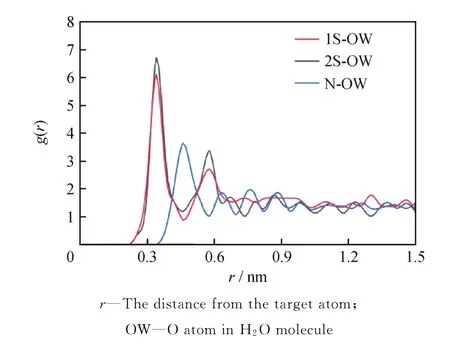
图8 表面活性剂亲水基团与H2 O径向分布函数Fig.8 Radial distribution functions of the surfactants hydrophilic groups with H2 O
—OSO3-和—SO3-附近Na+的径向分布函数分别用g1S-Na+(r)和g2S-Na+(r)表示。图9为表面活性剂亲水基团和H2O中Na+离子的径向分布函数。由图9可以看出,对于n(SDS)/n(SB12-3)=1/1体系中的Na+离子,g1S-Na+(r)和g2S-Na+(r)都存在明显的峰,Na+绝大多数吸附在紧靠极性头基的水中,只有极少量游离在水层中,Na+在界面的聚集浓度远大于水溶液中的浓度,这与不同摩尔比表面活性剂在油-水体系中的密度分布分析结果(见图3)相一致。当r=0.330 nm时,g1S-Na+(r)出现峰值,达到32.16;g2S-Na+(r)峰值(38.68)比g1S-Na+(r)的峰值大一点。这表明—SO3-与Na+之间有很强的静电效应,导致较多的Na+聚集在—SO3-附近,这是因为—OSO3-与SB12-3中的—N+离得较近,—N+与Na+产生静电斥力,所以导致Na+与—OSO3-离得较远,峰值较小。Na+与亲水头基的r值为0.330 nm,小于其与OW的r值(0.336 nm),表明Na+可以进入水化膜,会影响亲水基团和水分子的水化作用。
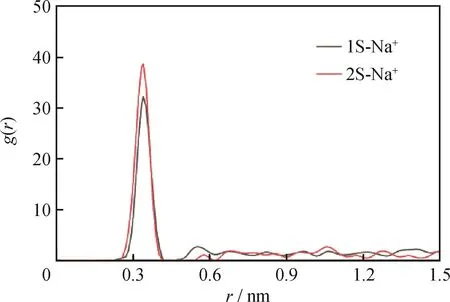
图9 表面活性剂亲水基团与Na+离子之间的径向分布函数Fig.9 Radial distribution functions between the hydrophilic groups of the surfactants and Na+ion
3 结 论
(1)采用分子动力学模拟方法,研究了SDS/SB12-3表面活性剂混合体系在油-水界面的聚集行为,发现当n(SDS)/n(SB12-3)=1/1时该混合体系的界面形成能达到最小(-640.959 kJ/mol)。
(2)SB12-3的—N+与SDS的—OSO3-静电相互作用导致界面厚度较大(1.942 nm),混合体系较稳定;由于SB12-3的—N+通过静电牵引接近SDS的—OSO3-,阻碍了Na+在—OSO3-附近的分布,因此—OSO3-与Na+径向分布函数峰值较小。
(3)SB12-3的—SO3-基团的氧原子优先与水形成氢键且相互作用最强;2种表面活性剂的头基与尾链的夹角和疏水尾链序参数结果表明,SDS烃链在油-水界面处更接近于界面法线(Z轴),其亲脂性更强。
