碱土及锰系复合氧化物担载Fe-K催化剂的费-托合成性能
2021-08-24刘振新赵晨曦贾高鹏田红美高玉集邢
刘振新赵晨曦贾高鹏田红美高玉集邢 宇
(郑州轻工业大学 材料与化学工程学院 河南省表界面科学重点实验室,河南 郑州 450002)
煤、天然气、生物质等富碳原料可以加工制备合成气(CO+H2)。费-托合成(Fischer-Tropsch synthesis)是指以合成气为原料,经过催化转化生成优质清洁能源和化工原料的过程,能够有效缓解对石油的依赖以及对环境的污染[1-4]。费-托合成的产物种类广泛,包括各种烃类化合物和其他各类有机含氧化合物,研究中通常以催化剂和反应条件的调控来提高目标产物的选择性[5-7]。
Mn和碱土族元素的氧化物均可作为固体碱改变催化剂的表面碱性。碱土金属由于其电子对供体性质以及其简单氧化物与载体氧化铝类似的某些特性,多用于费-托合成过程中催化剂的简单氧化物型助剂,能够在一定程度上抑制水煤气变换反应活性,加快烃类产物的生成速率[8-9]。Mg元素的施加能够提高催化剂的比表面积,增加催化剂表面碱性,进一步加快活性组分的还原和碳化,同时抑制水煤气变换反应并降低CO2选择性[10-12]。Ca元素的引入能够加速催化剂的碳化,增加催化剂表面碱性,从而抑制CO加氢速率,有效抑制甲烷的生成并提高烯/烷比;但Ca的过多施用会抑制催化剂的还原和碳化[13-15]。将Sr元素加入Fe基催化剂中,能够抑制O在Fe物种中的移动,有利于费-托反应过程中碳化物的生成,从而抑制催化剂的加氢作用,降低甲烷选择性的同时提高重质烃和烯烃的选择性[16-18]。Ba元素能够促进催化剂表面的CO解离吸附,相比于碱土金属Mg、Ca、Sr,催化剂活性有所提高,对于轻质烃的抑制作用更加明显,重质烃和烯烃选择性有所提高[18-20]。Mn元素能够提高Fe基催化剂的还原能力,提高Fe活性相的分散度,增加催化剂的催化活性,同时Fe、Mn之间的强相互作用能够抑制催化剂的失活,使催化剂在反应过程中更为稳定[21-22]。研究表明,在费-托反应过程中,适量Mn的加入能抑制甲烷的选择性,增加低碳烯烃和C5+的选择性[23-24]。
与简单氧化物相比,复合氧化物通常具有性质易于调变、熔点更高、耐热、耐水热等特点[25-30]。目前,对于碱土金属和Mn元素多用于费-托合成催化剂的助剂研究,作为费-托催化剂载体的研究则较少[31]。笔者早在2015年就报道了铝酸锌尖晶石载体对Co基费-托催化剂的影响,强调了尖晶石、钙钛矿等复合氧化物在费-托领域的巨大应用潜力[25]。近几年关于尖晶石在费-托反应中的应用研究层出不穷[32-37],尤其是尖晶石与沸石(如:丝光沸石[37]、AlPO4-18[35]、SAPO-34[32,34,36])的耦合在费-托合成制低碳烯烃领域取得了重大进展。然而,沸石的晶内孔易于受到催化剂积炭结焦的影响,且费-托过程涉及CO分子的C—O键解离、碳链增长甚至CO歧化,积炭结焦难以避免[38]。
尖晶石载体在费-托反应中的基础研究仍较少[26-27]。笔者在研究中发现,载体中具有尖晶石结构的复合氧化物Zn Al2O4相,在费-托反应过程中能够保持稳定结构[26-30],经过1200℃高温钝化,可以烧除提供表面酸性中心的表面羟基,使催化剂表面得以呈现适度的碱性,相比低煅烧温度更有利于生成低碳烯烃[26]。基于上述情况,为了探索Mn和碱土族等固体碱源潜在元素的相关催化性质和结构变化性质,笔者进行了Mn和碱土系复合氧化物担载Fe-K催化剂的合成气费-托转化基础性研究,制备了1200℃高温煅烧的含Mn或碱土族元素的复合氧化物固体碱载体,担载活性金属Fe和K助剂后制成Fe-K2O/MAl2O4(M为Mg、Ca、Sr、Ba、Mn)费-托催化剂,对其物理化学性质进行表征,比较其CO加氢性能。
1 实验部分
1.1 原料和试剂
硝酸锰(Mn(NO3)2(质量分数50%水溶液)、硝酸钙(Ca(NO3)2·4 H2O)、硝酸锶(Sr(NO3)2)、硝酸钡(Ba(NO3)2)、碳酸氢钠(Na HCO3)、碳酸钾(K2CO3),均为分析纯,国药集团化学试剂有限公司产品;硝酸镁(Mg(NO3)2·6 H2O)、硝酸铝(Al(NO3)3·9H2O)、柠檬酸铁铵,均为分析纯,上海阿拉丁生化科技股份有限公司产品。
1.2 MAl2 O4载体和Fe-K2 O/MAl2 O4催化剂的制备
1.2.1 MAl2O4载体的制备
MAl2O4载体的制备方法为共沉淀-挤条干燥-煅烧法[31]:预制备30 g载体,以制备Ba Al2O4型载体为例,首先按Al/Ba摩尔比(n(Al)/n(Ba))为2称取Al(NO3)3·9H2O和Ba(NO3)2于烧杯中,加入去离子水搅拌混合溶解至1000 m L;称取沉淀Al、Ba元素所需当量1.5倍(即过量50%)的沉淀剂Na HCO3于烧杯中,加入去离子水搅拌溶解至1000 m L;在温度40℃和转速800 r/min条件下将上述2种溶液以相同的滴加速率同步滴加,实施共沉淀,同步滴加完毕后继续在40℃搅拌陈化1 h;抽滤后使用总计6000 m L的去离子水多次充分洗滤。在80℃下将滤饼置于鼓风干燥箱中部分脱水后,称取1.5 g(预制备载体质量的5%)的田菁胶粉(做为助挤剂使用)与半干的滤饼混匀、捏合1 h后挤条,自然阴干,再于110℃下干燥2 h,之后将所得样品在空气气氛下于1200℃下煅烧24 h,得到含Al/Ba元素的BaAl2O4型载体。Mg Al2O4、CaAl2O4、Sr Al2O4型载体的制备过程均与上述制备Ba Al2O4型载体的步骤相同。
MnAl2O4型载体的制备流程不同于其他载体。其制备方法的特点是用0.3487 mol的Al(NO3)3·9H2O溶液与沉淀剂Na HCO3(1.5689 mol)溶液发生沉淀,将过滤后的滤饼脱水干燥后,按n(Al)/n(Mn)=2加入0.1743 mol的Mn(NO3)2,再加入1.5 g的田菁胶粉,之后制备过程与上述其他催化剂载体的相同,最后得到Mn Al2O4型载体。
1.2.2 Fe-K2O/MAl2O4催化剂的制备
以制备Fe-K2O/Ba Al2O4催化剂为例,按照m(Fe)∶m(K2O)∶m(Ba Al2O4)=15∶2∶83的比例,以使用30 g的Ba Al2O4型载体计,称取0.0971 mol的柠檬酸铁铵和0.0077 mol的K2CO3(使用K2CO3的催化剂组成中一般以K2O质量分数来表示K含量),等体积浸渍入BaAl2O4型载体后,于空气气氛下在350℃煅烧4 h,最终得到氧化态的Fe-K2O/Ba Al2O4催 化 剂。Mg Al2O4、CaAl2O4、Sr Al2O4、Mn Al2O4型载体担载Fe、K元素的催化剂制备过程与此相同。
1.3 MAl2 O4载体和Fe-K2 O/MAl2 O4催化剂的表征
采用Panalytical X’Pert Pro X射线衍射仪测定所制备载体和催化剂的物相结构。采用Perkin-Elmer Elan 9000型电感耦合等离子体质谱仪测定元素含量。采用Quantach rome NOVA1000表面积与孔径分析仪测定N2物理吸附介孔分布。采用Quantachrome ChemStarTM化学吸附分析仪(Chem-Bet检测器)进行CO2程序升温脱附(CO2-TPD),催化剂装填量100 mg,检测器电流127.0 m A,催化剂首先通过550℃原位H2的预还原,继而冷却至50℃时,使用CO2-Ar混合气实施CO2吸附,之后在50℃下用氦气吹扫去除物理吸附的CO2,然后再以升温速率10℃/min,从50℃升温到800℃进行CO2-TPD检测。采用美国FEI公司QUANTA Q400热场发射电子显微镜观察催化剂的微观形貌。
1.4 Fe-K2 O/MAl2 O4催化剂的费-托合成反应催化性能评价
采用天津市天大北洋化工实验设备有限公司生产的固定床管式费-托合成反应器,进行合成气转化反应。将4 m L制得的催化剂(40~60目)装入反应管中,先在常压下于550℃用50%H2-50%He(体积分数)混合气原位还原6 h,继而切换至预混合的合成气(nH2∶nCO∶nAr=45∶45∶10)并缓慢升压至2.0 MPa,以升温速率2℃/min升温至催化剂的活性反应温度进行合成气催化转化反应,空速为1500 mL/(g cat·h)。反应过程中合成气单程通过反应器。反应后降至室温(约30℃)和常压(0.1 MPa),用高纯氩气吹扫3 h后,收集反应后的催化剂。
氩气作为合成气的内标组分用于气相色谱分析。采用北京普瑞分析仪器有限公司生产的GC-5890型气相色谱仪测定CO、CH4和CO2,TCD检测器配有TDX-01填充柱(长度3 m)。采用北京普瑞分析仪器有限公司生产的GC-6890型气相色谱仪进行烃类分析,FID检测器配有PLOT Al2O3/S(50 m×0.53 mm×25μm)毛细管柱。
CO转化率(xCO)为已转化的CO物质的量(nCO,mol)占引入反应器内的CO物质的量(n'CO,mol)的百分比率,其计算式如式(1)所示。
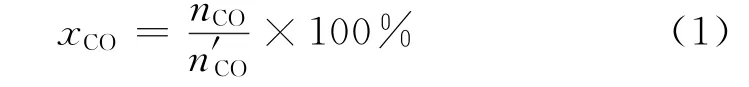
CO2选择性(sCO2)为CO转化为CO2的物质的量(n″CO,mol)占已转化的CO物质的量(nCO,mol)的百分比率,其计算式如式(2)所示。

烃类选择性(sHC)为C1(即甲烷)、C2~C4、C5+等烃类(HC)产物分别在全部烃类产物中所占的碳基(即基于C元素)物质的量的百分比率。全部烃类产物包括全部烷烃和烯烃(产物中未发现炔烃)。如:C2=~C4=的烃类选择性(sHC(C2=-C4=))即为C2~C4烃类产物中烯烃在全部烃类产物中所占的碳基物质的量的百分比率,其计算式如式(3)所示。
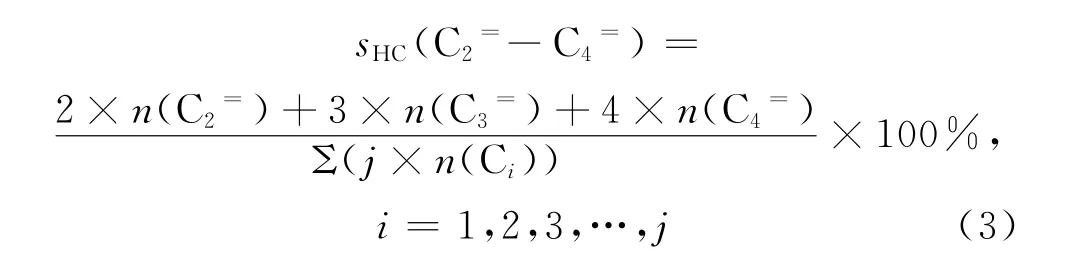
烯/烷比(nO/nP)为烃类产物中某碳数烯烃所含碳基物质的量与同碳数烷烃所含碳基物质的量的比值(摩尔比)。如:C2~C4烯/烷比为烃类产物中C2~C4烯烃(包括C2=、C3=、C4=)所含碳基物质的量与C2~C4烷烃(包括C20、C30、C40)所含碳基物质的量的比值,其计算式如式(4)所示。
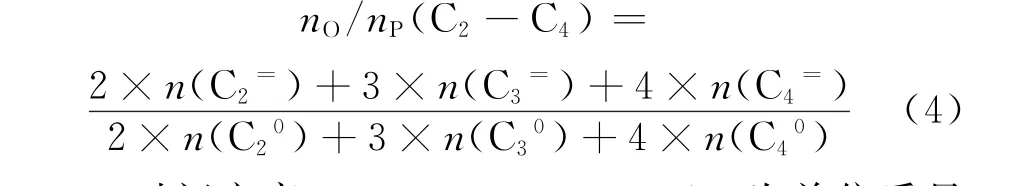
Fe时间产率(yFe,t,iron time yield)为单位质量的Fe(mFe)在单位时间(t)内将CO分子转化为烃类产物的碳基物质的量,mmolCO/(gFe·s),其计算式如式(5)所示。
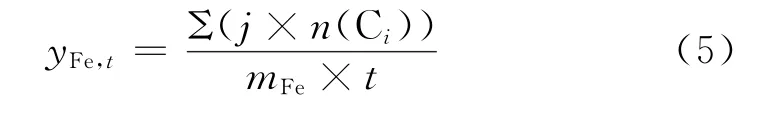
2 结果与讨论
2.1 MAl2 O4载体和Fe-K2 O/MAl2 O4催化剂的结构特征
图1为MAl2O4(M为Mg、Ca、Sr、Ba、Mn)载体以及费-托反应前后Fe-K2O/MAl2O4催化剂的X射线衍射(XRD)谱图。由图1(a)可知:Mg Al2O4型载体的结晶态物相主要是Mg Al2O4(JCPDS:21-1152)以及少量的β-Al2O3(JCPDS:10-0414)[31];Ca Al2O4型载体的结晶态物相包括Ca Al2O4(JCPDS:23-1036)和Ca Al4O7(JCPDS:23-1037);Sr Al2O4型载体的结晶态物相包括Sr Al2O4(JCPDS:34-0379)和Sr Al4O7(JCPDS:25-1289);Ba Al2O4型载体的结晶态物相主要是Ba Al2O4(JCPDS:17-0306)以及微量的Ba2Al10O17(JCPDS:48-442);Mn Al2O4型载体的结晶态物相包括Mn Al2O4(JCPDS:29-0880)、Mn5O8(JCPDS:39-1218)和α-Al2O3(JCPDS:10-0173)。
由图1(b)可知:Fe-K2O/Mg Al2O4催化剂的结晶态物相主要是Fe0(JCPDS:06-0696)和Mg Al2O4以及少量的β-Al2O[31]3;Fe-K2O/Ca Al2O4催化剂的结晶态物相主要为接近Ca2Fe2O5(JCPDS:47-1744)的铁酸钙和Fe0,其铝物种为X射线无定形状态;Fe-K2O/Sr Al2O4催化剂的结晶态物相主要为接近Sr4Al14O25(JCPDS:52-1876)的铝酸锶和Fe0;Fe-K2O/Ba Al2O4催化剂的结晶态物相包括Ba Al2O4、Fe0以及接近Ba2Al10O17(JCPDS:48-442)的铝酸钡;Fe-K2O/Mn Al2O4催化剂的结晶态物相包括Fe0、Mn Al2O4、Mn5O8、α-Al2O3以及接近K4MnO4(JCPDS:31-1051)的锰酸钾。
由图1(c)可知:反应后的Fe-K2O/Mg Al2O4催化剂,其结晶相包括β-Al2O3、Mg Al2O4和Fe5C2(JCPDS:51-0997),说明该催化剂中的Fe0相在CO加氢反应过程中碳化生成了Fe5C2活性相;反应后的Fe-K2O/Ca Al2O4催化剂,其结晶态物相包括接近Ca4Fe9O17(JCPDS:17-0105)的铁酸钙和Fe5C2,其铝物种仍为X射线无定形状态;反应后的Fe-K2O/Sr Al2O4催化剂,其结晶态物相包括接近Sr4Al14O25(JCPDS:52-1876)的铝酸锶和Fe5C2;反应后的Fe-K2O/Ba Al2O4催化剂,其结晶态物相包括接近Al3Fe5O12(JCPDS:49-1657)的铁酸铝、Ba Al2O4以及接近Ba2Al10O17(JCPDS:48-442)的铝酸钡,没有观察到活性相Fe5C2的衍射峰(这可能是由于大部分Fe元素因生成铁酸铝而被消耗,残余Fe元素所生成的Fe5C2数量过少而难以形成目测可见的XRD衍射峰);反应后的Fe-K2O/Mn Al2O4催化剂,其结晶态物相包括Mn Al2O4、α-Al2O3、Fe5C2以及类似(Mn0.983Fe0.017)2O3(JCPDS:31-1051)的锰(铁)氧化物。
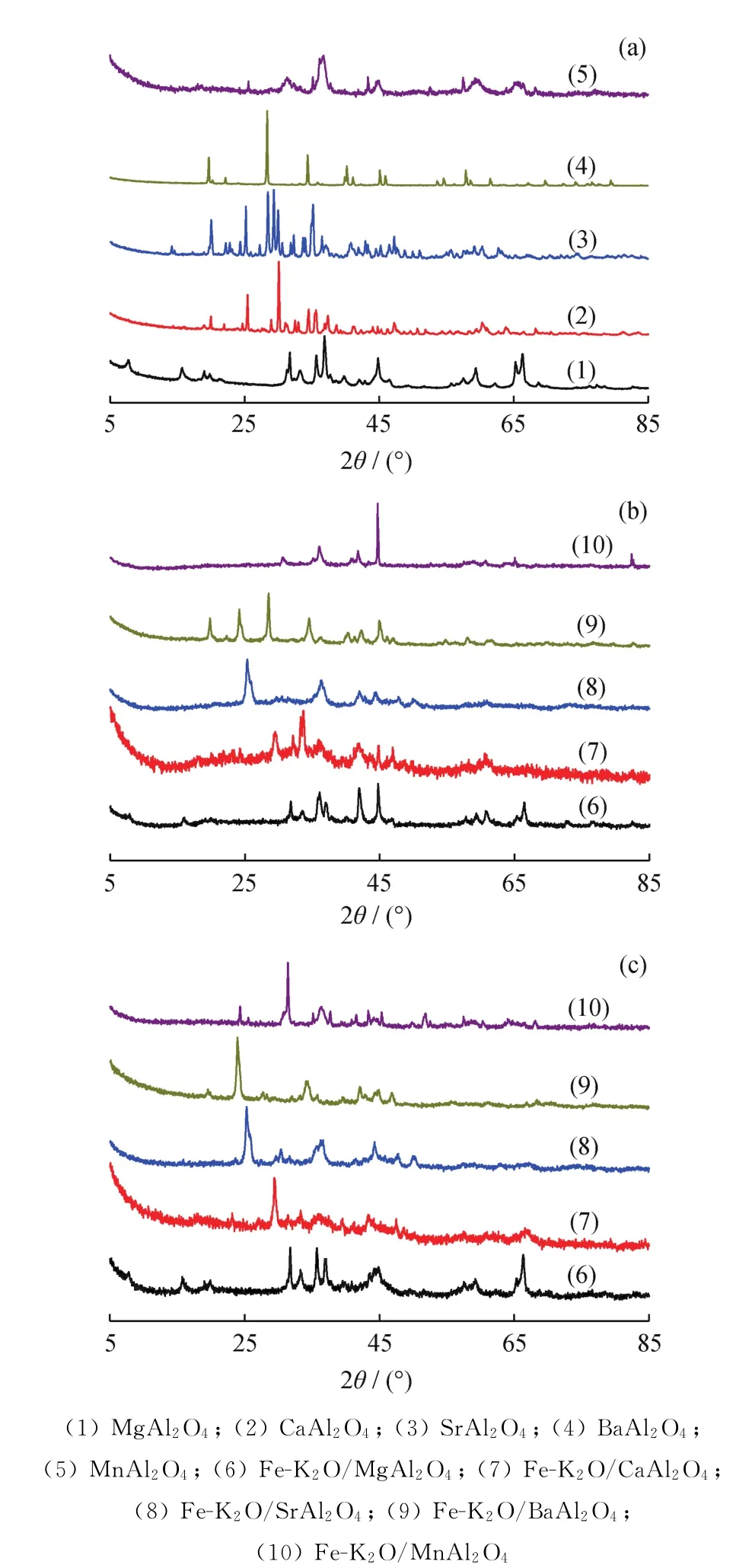
图1 MAl2 O4载体和费-托反应前后Fe-K2 O/MAl2 O4催化剂的X射线衍射谱图Fig.1 X-ray diffraction patterns of MAl2 O4 supports,Fe-K2 O/MAl2 O4 catalysts before and after Fischer-Tropsch(F-T)reactions
2.2 Fe-K2 O/MAl2 O4催化剂的孔织构特征
图2为费-托反应前Fe-K2O/MAl2O4催化剂的N2物理吸附结果。由图2(a)可知,所制备的5种新鲜还原态Fe-K2O/MAl2O4催化剂均归属于Type IV型等温线,证明这5种催化剂均属于介孔吸附剂[39]。它们的滞后环在形态上均与H3型滞后环相似,说明这5种催化剂的介孔主要是由板状/片状颗粒构筑而成的狭缝形孔隙[39]。Fe-K2O/Mg Al2O4、Fe-K2O/Ca Al2O4和Fe-K2O/Sr Al2O4的BET比表面积接近,分别为17.9、22.6、20.1 m2/g,处于相对较高区间,说明耐烧结性能相对较好;Fe-K2O/Ba Al2O4和Fe-K2O/Mn Al2O4的BET比表面积接近,分别为8.6、5.0 m2/g,处于相对较低区间,说明耐烧结性能相对较差。Fe-K2O/Mg Al2O4、Fe-K2O/Ca Al2O4和Fe-K2O/Ba Al2O4的BET平均孔直径接近,分别为46.2、39.0、40.6 nm,处于相对较高值域;Fe-K2O/Sr Al2O4和Fe-K2O/Mn Al2O4的平均孔直径接近,分别为23.4、22.2 nm,处于相对较低值域。Fe-K2O/MgAl2O4和Fe-K2O/CaAl2O4的总孔体积相对较高,分别达到0.21、0.22 cm3/g;Fe-K2O/Sr Al2O4、Fe-K2O/Ba Al2O4和Fe-K2O/Mn Al2O4的总孔体积逐次降低,分别为0.12、0.09、0.03 cm3/g。
第一主孔径是指dV/dD最高的主孔径,第二主孔径和第三主孔径则是dV/dD逐次降低的主孔径。图2(b)说明,Fe-K2O/CaAl2O4和Fe-K2O/BaAl2O4均有3个主孔径(前者分别位于16.1、1.3、3.8 nm;后者分别位于1.3、29.9、3.8 nm),且孔分布形态相似;Fe-K2O/Mg Al2O4和Fe/K2O/Sr Al2O4均有2个主孔径(前者分别位于1.5、29.8 nm;后者分别位于16.3、3.8 nm),但孔分布形态有所不同;Fe-K2O/Sr Al2O4的孔分布形态与Fe-K2O/Ca Al2O4和Fe-K2O/Ba Al2O4相似;Fe-K2O/Mn Al2O4有2个主孔径(分别位于1.8、2.4 nm),但孔分布形态与碱土族的4种催化剂完全不同。
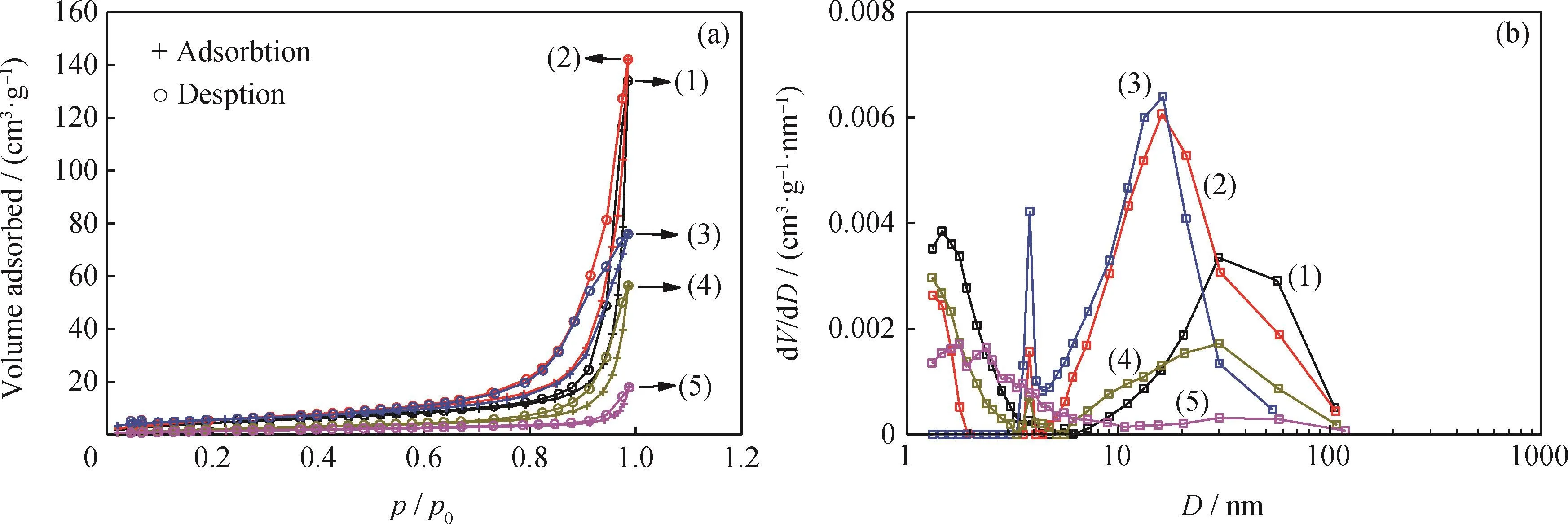
图2 费-托反应前Fe-K2 O/MAl2 O4催化剂的N2物理吸附结果Fig.2 N2 sorption measurement results of Fe-K2 O/MAl2 O4 catalysts before Fischer-Tropsch reactions
2.3 Fe-K 2 O/MAl2 O4催化剂的微观形貌
图3为费-托合成反应前Fe-K2O/MAl2O4催化剂的微观形貌照片。由图3(a)和(b)可见,Fe-K2O/Mg Al2O4催化剂由片状和块状2种颗粒构成,其中片状颗粒为主,块状颗粒为辅;片状颗粒为二次颗粒,由粒径20~30 nm的一次颗粒聚集而成;块状颗粒的边长可达约1μm。由图3(c)可见,Fe-K2O/Ca Al2O4催化剂主要是粒径约50 nm的一次颗粒板结而成的二维板壳体,也有一些不规则形状的微米/亚微米团聚体。图3(d)表明,Fe-K2O/Sr Al2O4催化剂主要是不规则形态的片层状二次颗粒,边长在亚微米/微米级;构成这些片层状二次颗粒的是近似球形的粒径20~30 nm的一次颗粒。图3(e)表明,Fe-K2O/Ba Al2O4催化剂主要是由粒径30~40 nm的一次颗粒团聚而成的亚微米二次颗粒。图3(f)表明,Fe-K2O/Mn Al2O4催化剂主要是形状不规则的亚微米/微米片状体,以及粒径30~50 nm的一次颗粒的沉积态聚集体。

图3 费-托反应前Fe-K2 O/MAl2 O4催化剂的FESEM照片Fig.3 FESEM images of Fe-K2 O/MAl2 O4 catalysts before Fischer-Tropsch reactions
2.4 Fe-K 2 O/MAl2 O4催化剂的表面碱度
Fe-K2O/MAl2O4催化剂的表面碱度用CO2气体实施化学吸附-程序升温脱附(CO2-TPD)测定。图4为经过原位还原的Fe-K2O/MAl2O4催化剂的CO2-TPD曲线。表1为Fe-K2O/MAl2O4催化剂的CO2脱附峰相应的积分数据。5种催化剂的CO2-TPD曲线在形态上都是左低右高(Ihigh/Ilow均显著高于1.0),其高温脱附峰的积分面积也都显著大于低温脱附峰,表明5种催化剂的表面碱性吸附位在数量上均以弱碱位为辅,以强碱位为主。同时,每种催化剂的高温脱附峰面积百分比、高温脱附峰温度均有差异,说明不同的组成元素(Mg、Ca、Sr、Ba、Mn)对于催化剂的表面碱度分布具有显著影响。
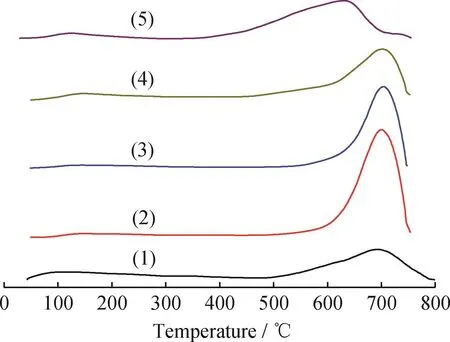
图4 费-托反应前Fe-K2 O/MAl2 O4催化剂的CO2程序升温脱附Fig.4 CO2 temperature-programmed desorption of Fe-K2 O/MAl2 O4 catalysts before Fischer-Tropsch reactions
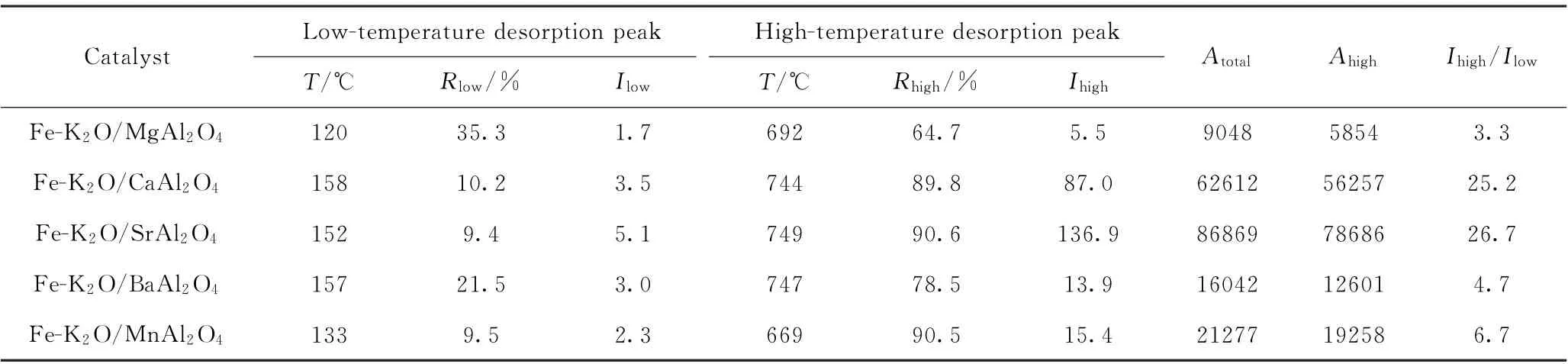
表1 Fe-K2 O/MAl2 O4催化剂的CO2-TPD的积分数据Table 1 Integral data of CO2-TPD results of Fe-K2 O/MAl2 O4 catalysts
Fe-K2O/Sr Al2O4催化剂有2个CO2脱附峰:位于152℃的低温脱附峰和位于749℃的高温脱附峰。CO2脱附峰的对应温度越高,说明作为酸性分子的CO2在催化剂表面碱性位上的化学吸附越牢固(即越难以脱附),证明该吸附位的表面碱性越强。峰面积越大,说明该种碱性位数量越多;峰面积越小,则说明该种碱性位数量越少。Fe-K2O/Sr Al2O4催化剂的低温峰面积占比(Rlow)仅为9.4%,而高温峰面积占比(Rhigh)高达90.6%,说明其表面强碱位明显多于弱碱位。综合XRD结构分析和CO2-TPD数据来看,Fe-K2O/Sr Al2O4催化剂的749℃高温脱附峰很可能来源于该样品中铝酸锶相和钾物种的共同作用,而在152℃的低温脱附峰则很可能来源于该样品中的Fe0相。Fe-K2O/Mg Al2O4、Fe-K2O/CaAl2O4、Fe-K2O/BaAl2O4、Fe-K2O/Mn Al2O4的CO2-TPD曲线可以依此类推解析。
如前所述,在MAl2O4载体和费-托反应前后Fe-K2O/MAl2O4催化剂的XRD谱图中,没有发现各个载体相含有任何结晶态碳酸盐。此外,Fe-K2O/MAl2O4催化剂制备中添加的K2CO3助剂,热分解虽能释放CO2气体,但K2CO3在常压下的热分解起始温度位于其熔点891℃[40],因此不会对50~800℃的CO2-TPD结果产生有效影响。
2.5 Fe-K2 O/MAl2 O4催化剂的费-托合成反应催化性能评价结果
2.5.1 Fe-K2O/MAl2O4催化剂的催化活性
图5为不同反应温度下Fe-K2O/MAl2O4催化剂的费-托合成反应催化活性。在图5中,通过5种催化剂CO转化率与反应温度关系曲线与CO转化率为50%水平线的相交点,拟合出5种催化剂在CO转化率为50%时对应的反应温度,该反应温度通常用来比较催化剂活性,即该反应温度越低,说明催化剂活性越高;反之亦然。因此可以得出,Fe-K2O/MAl2O4催化剂的费-托合成反应活性由高到低的次序为Fe-K2O/Sr Al2O4、Fe-K2O/BaAl2O4、Fe-K2O/CaAl2O4、Fe-K2O/Mg Al2O4、Fe-K2O/Mn Al2O4。对 碱 土 族元素来说,这一次序与元素周期表中的顺序并不完全一致,说明对催化剂活性有影响的因素是复杂的。表1和图4中的CO2-TPD高温脱附峰温度由高到低的次序也是Fe-K2O/Sr Al2O4、Fe-K2O/Ba Al2O4、Fe-K2O/CaAl2O4、Fe-K2O/MgAl2O4、Fe-K2O/MnAl2O4,这说明催化剂表面碱性位的碱性强度对催化剂的活性有非常重要的影响,表面碱性位的碱性越强则催化剂的活性越高,这同时也表明催化剂表面碱性位的碱性越强就会对CO和H2分子产生更强的活化作用,导致更高的CO转化率。反应速率取决于CO分子的解离活化[41]。当CO分子吸附于表面Fe物种上时,CO分子倾向于从Fe上拉电子[42]。较强的碱(如K2O等),作为电子供体,可以为Fe—C键供电子,增强Fe—C键,削弱C—O键,使得C—O键更易被H2攻击,即可以促进CO在Fe上的吸附与解离活化,从而增加费-托反应速率[42]。这很可能就是表面碱性位碱性最强的Fe-K2O/Sr Al2O4催化剂具有最高催化活性的原因。将来拟在Sr含量调整和对照表征方面对Fe-K2O/Sr Al2O4催化剂开展进一步的研究。
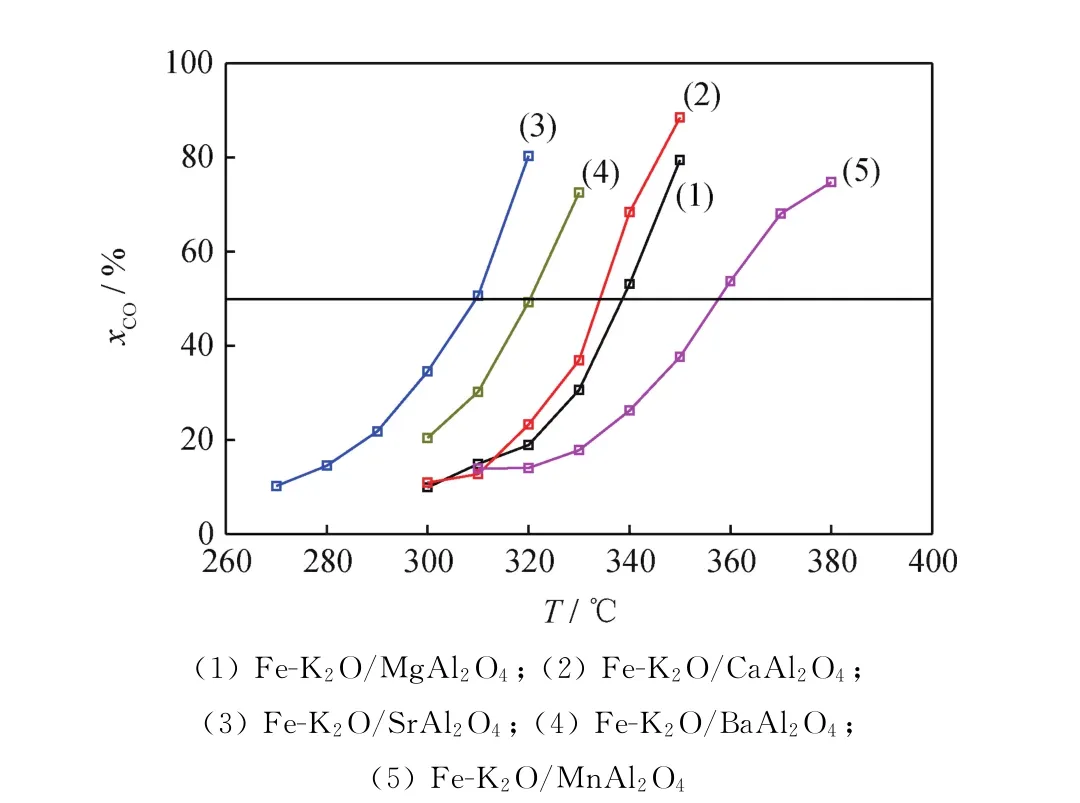
图5 不同反应温度(T)下Fe-K2 O/MAl2 O4催化剂的费-托合成反应催化活性Fig.5 Catalytic activity of Fe-K2 O/MAl2 O4 catalysts for Fischer-Tropsch synthesis reaction at different reaction temperatures(T)
5种Fe-K2O/MAl2O4催化剂的费-托合成反应催化性能列于表2~6。由表2~6可知,5种催化剂的CO2选择性均不超过50%,处于费-托合成制烯烃催化剂的正常范围(即≤50%)[38]。
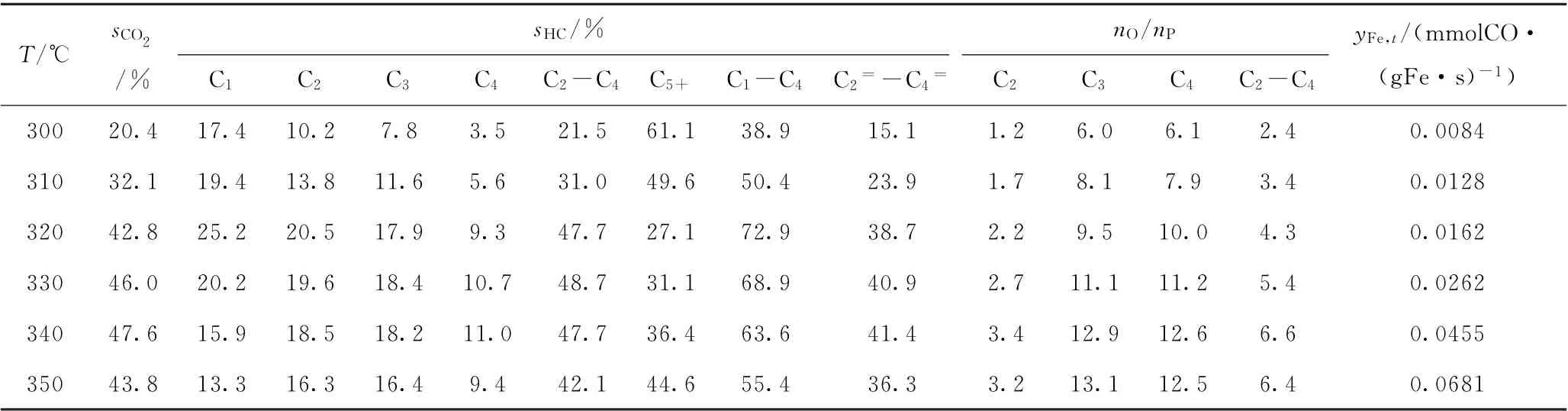
表2 Fe-K2 O/MgAl2 O4催化剂的费-托合成催化性能Table 2 Fischer-Tropsch synthesis performance of Fe-K2 O/MgAl2 O4 catalyst
2.5.2 Fe-K2O/MAl2O4催化剂的抑制加氢能力
还原态催化剂的抑制加氢能力体现在烃类产物的烯/烷比上,抑制加氢的能力越强,则烯/烷比越高,即烃类产物中烯烃的占比越高,而与之相应的烃类产物中烷烃的占比越低。图6为不同反应温度和CO转化率下Fe-K2O/MAl2O4催化剂费-托合成反应的抑制加氢能力。
由图6可见,随着反应温度增加,CO转化率相应增加,Fe-K2O/Sr Al2O4催化剂的C2~C4烯/烷比呈下降趋势,由4.3降至1.4。费-托合成属于加氢反应,是放热的,从热力学上讲,升温不利于放热的加氢反应,因此从理论上看升温本应该导致烯/烷比升高而非降低。另一方面,费-托反应包含一次反应和二次反应,一次反应中CO加氢产生的是烯烃而非烷烃,二次反应中烯烃可以进一步深度加氢生成烷烃[43]。因此,Fe-K2O/Sr Al2O4催化剂的C2~C4烯/烷比随反应温度的提高呈下降趋势,说明随着反应温度的提高,Fe-K2O/Sr Al2O4催化剂明显发生了二次加氢反应。如图6(b)所示,5种Fe-K2O/MAl2O4催化剂中,除Fe-K2O/Sr Al2O4外,其他4种催化剂的烯/烷比变化趋势均是先升后降,说明反应温度对催化剂二次加氢反应的引发作用是有差异的,Fe-K2O/Sr Al2O4催化剂的二次加氢反应相对较容易被反应温度的升高所引发,其他4种催化剂的二次加氢反应则相对较难被引发。由于5种Fe-K2O/MAl2O4催化剂的活性不同,反应温度区间存在明显差异,因此,5种催化剂二次加氢反应被反应温度升高所引发的由易到难次序为Fe-K2O/Sr Al2O4、Fe-K2O/CaAl2O4、Fe-K2O/BaAl2O4、Fe-K2O/Mg Al2O4、Fe-K2O/Mn Al2O4。这一次序与5种催化剂CO2-TPD高温脱附峰温度由高到低的次序基本一致,说明催化剂表面碱性位的碱性越强则越容易被反应温度的升高引发二次加氢反应。
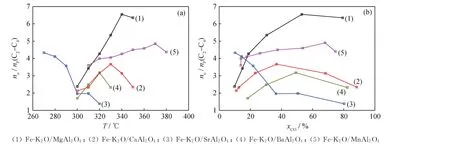
图6 不同反应温度(T)和CO转化率(x CO)下Fe-K2 O/MAl2 O4催化剂费-托合成反应的抑制加氢能力Fig.6 Hydrogenation inhibition ability of Fe-K2 O/MAl2 O4 catalysts for Fischer-Tropsch synthesis at different reaction temperatures(T)and CO conversions(x CO)
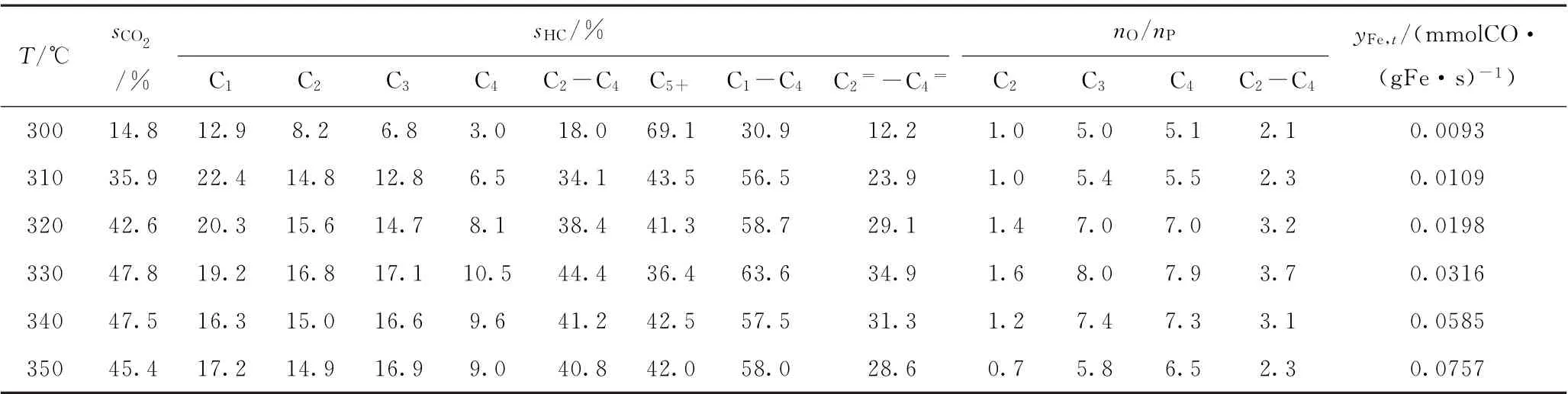
表3 Fe-K2 O/CaAl2 O4催化剂的费-托合成催化性能Table 3 Fischer-Tropsch synthesis performance of Fe-K2 O/CaAl2 O4 catalyst
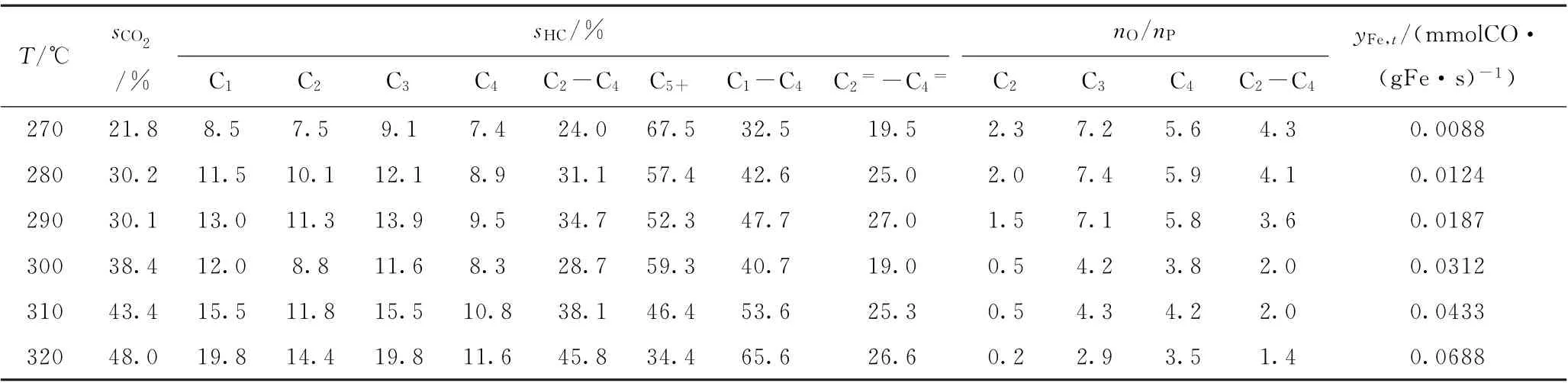
表4 Fe-K2 O/Sr Al2 O4催化剂的费-托合成催化性能Table 4 Fischer-Tropsch synthesis performance of Fe-K2 O/Sr Al2 O4 catalyst
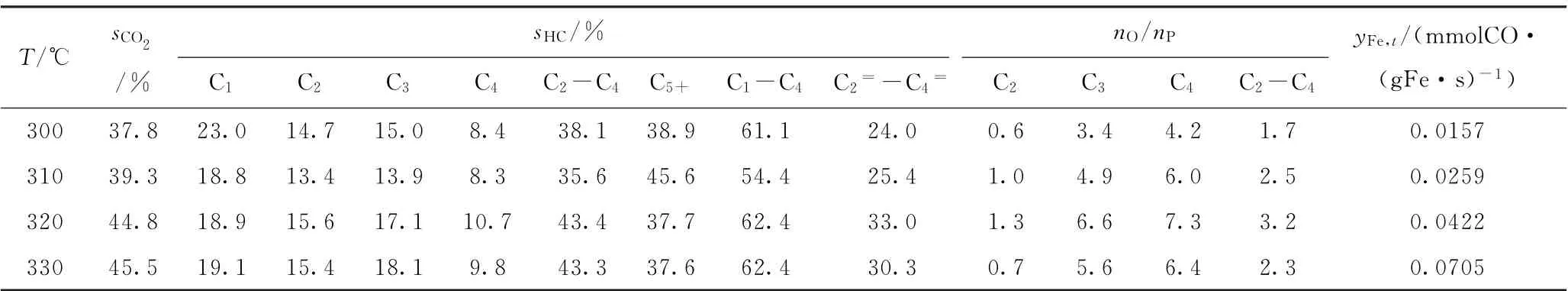
表5 Fe-K2 O/BaAl2 O4催化剂的费-托合成催化性能Table 5 Fischer-Tropsch synthesis performance of Fe-K2 O/BaAl2 O4 catalyst
由于不同催化剂的费-托反应活性差异很大,如Fe-K2O/Sr Al2O4催化剂在320℃的转化率即已高达80.3%,而Fe-K2O/Mn Al2O4催化剂在380℃的转化率才达到74.7%,各催化剂反应温度区间的共同交集较少,因此选择反应温度为310℃进行催化性能比较。310℃是共有反应温度,而且可以避免出现过高的CO转化率,因为费-托合成反应通常会避免过高的转化率,从而避免产生过高的水蒸气分压(水蒸气分压过高会导致催化剂加速失活)。由表2~6可见,在310℃时,5种Fe-K2O/MAl2O4催化剂的C2~C4烯/烷比由高到低的次序为Fe-K2O/Mn Al2O4(3.6)、Fe-K2O/Mg Al2O4(3.4)、Fe-K2O/Ba Al2O4(2.5)、Fe-K2O/Ca Al2O4(2.3)、Fe-K2O/Sr Al2O4(2.0)。表1和图4中的CO2-TPD高温脱附峰温度由低到高的次序是Fe-K2O/MnAl2O4、Fe-K2O/MgAl2O4、Fe-K2O/CaAl2O4、Fe-K2O/BaAl2O4、Fe-K2O/Sr Al2O4。这些数据表明,5种催化剂的C2~C4烯/烷比与CO2-TPD高温脱附峰温度之间大致上呈反向关系,即5种催化剂的抑制加氢能力与其表面碱性位的碱性强度大致上呈反向关系。
2.5.3 Fe-K2O/MAl2O4催化剂的C-C偶合能力
C1~C4(即较短链烃类)的烃类选择性与C5+(即较长链烃类)的烃类选择性之和等于100%。理论上讲,催化剂的C-C偶合能力越强,则越易于生成较长链的烃类,即会导致C5+的烃类选择性越高、C1~C4的烃类选择性越低。
由表2~6可见,在310℃时,5种催化剂的C5+烃类选择性由高到低的次序为Fe-K2O/MnAl2O4(57.8%)、Fe-K2O/Mg Al2O4(49.6%)、Fe-K2O/Sr Al2O4(46.4%)、Fe-K2O/BaAl2O4(45.6%)、Fe-K2O/Ca Al2O4(43.5%)。前两者的CO2-TPD高温脱附峰温度均较低,处于669~692℃;后三者的CO2-TPD高温脱附峰温度均较高,处于744~749℃。后三者的CO2-TPD高温脱附峰温度都很接近,其C5+烃类选择性也较接近。这些数据表明,催化剂的C-C偶合能力与CO2-TPD高温脱附峰温度之间大致上呈反向关系,即5种催化剂的C-C偶合能力与其表面碱性位的碱性强度大致上呈反向关系。
一般来讲,随着反应温度的逐渐升高,反应中间体会加速从催化剂表面脱附,从而抑制C-C偶合过程,导致C5+烃类选择性的逐渐下降和C1~C4烃类选择性的逐渐上升。图7为不同反应温度和CO转化率下催化剂费-托合成反应的C-C偶合能力。由图7可知,除Fe-K2O/Sr Al2O4催化剂的C5+烃类选择性趋势与常规费-托催化剂(如Fe/Mn/K催化剂[7])较为接近外,其余4个催化剂的C-C偶合能力均显著不同于常规费-托催化剂[7]。由图7(b)可知,在CO转化率处于10%~20%的相对低位区间,Fe-K2O/Mn Al2O4、Fe-K2O/Mg Al2O4和Fe-K2O/Ca Al2O4的C5+烃类选择性均出现了近乎垂直的急速降低;在CO转化率处于20%以上的相对高位区间,4个催化剂的C5+烃类选择性基本呈上升或平稳趋势。这些数据表明,催化剂载体可以对Fe-K催化剂的C-C偶合能力产生重大影响,使Fe-K2O/Mn Al2O4、Fe-K2O/Mg Al2O4和Fe-K2O/Ca Al2O4催化剂在各自的较低反应温度区间(对应着各自的较低CO转化率区间)可以特别有效地遏制C-C偶合,使C5+烃类选择性急剧下降;但是随着反应温度的进一步升高,这些催化剂遏制C-C偶合的特性被逐渐削弱,导致C5+烃类选择性逐渐平稳甚至上升。Fe-K2O/Mn Al2O4催化剂在380℃时的CO转化率达到74.7%(见表6),再次出现了C5+烃类选择性的显著下降,说明对于该催化剂来说,从380℃时开始显现了反应温度对C-C偶合能力的有效抑制。
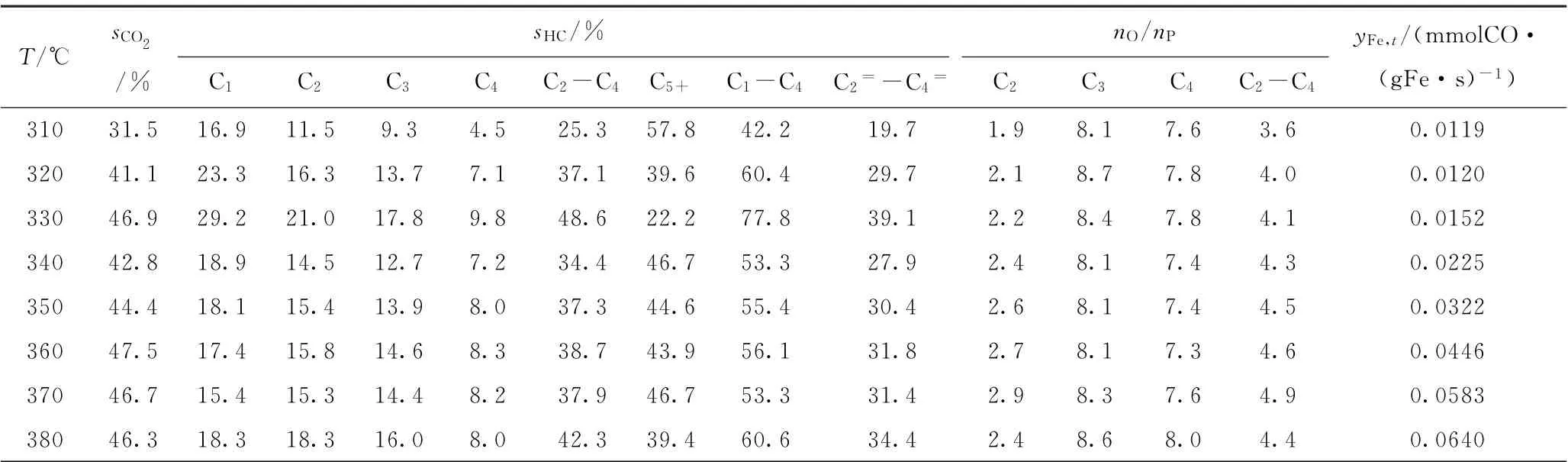
表6 Fe-K2 O/Mn Al2 O4催化剂的费-托合成催化性能Table 6 Fischer-Tropsch synthesis performance of Fe-K2 O/Mn Al2 O4 catalyst
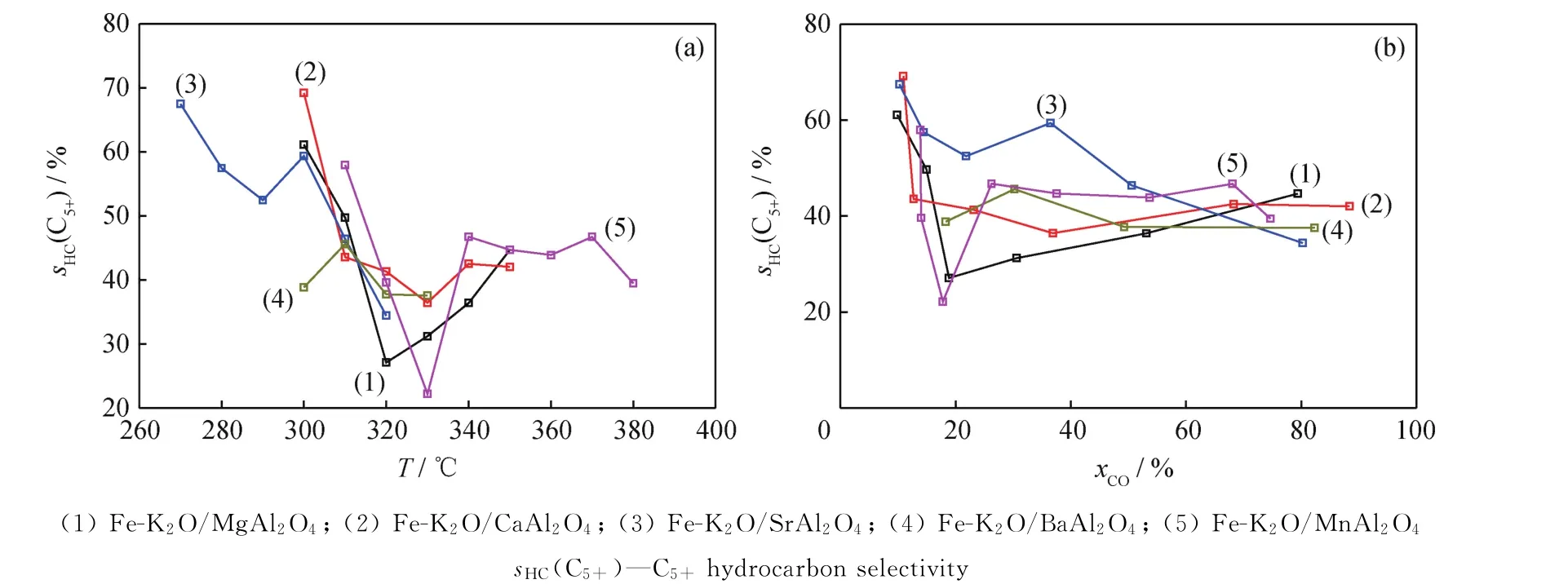
图7 不同反应温度(T)和CO转化率(x CO)下催化剂费-托合成反应的C-C偶合能力Fig.7 C-C coupling capability of Fe-K2 O/MAl2 O4 catalysts for Fischer-Tropsch synthesis at different reaction temperatures(T)and different CO conversions(x CO)
2.5.4 Fe-K2O/MAl2O4催化剂的C2=~C4=烃类选择性
由于各Fe-K2O/MAl2O4催化剂活性、抑制加氢能力、C-C偶合能力的差异,其C2=~C4=的烃类选择性最大值出现在不同的反应温度。5种Fe-K2O/MAl2O4催化剂的C2=~C4=烃类选择性最大值由高到低的次序为Fe-K2O/Mg Al2O4(41.4%)、Fe-K2O/Mn Al2O4(39.1%)、Fe-K2O/Ca Al2O4(34.9%)、Fe-K2O/Ba Al2O4(33.0%)、Fe-K2O/Sr Al2O4(27.0%)。这些数据表明,催化剂低碳烯烃选择性的高低与CO2-TPD高温脱附峰温度之间大致上呈反向关系,即高温脱附峰温度越高则催化剂低碳烯烃选择性会越低。也就是说,CO2-TPD高温脱附峰温度相对较低(669~692℃)的Fe-K2O/MgAl2O4、Fe-K2O/Mn Al2O4,比CO2-TPD高温脱附峰温度相对较高(744~749℃)的Fe-K2O/CaAl2O4、Fe-K2O/Sr Al2O4、Fe-K2O/BaAl2O4,更有利于促使反应生成低碳烯烃。因此,为了富产低碳烯烃,催化剂表面碱性位的碱性强度要适度(即CO2-TPD高温脱附峰温度不宜高于700℃)。
3 结 论
(1)制备了Fe-K2O/MAl2O4(M为Mg、Ca、Sr、Ba、Mn)复合氧化物型Fe基催化剂,用于合成气的费-托转化。5种催化剂均为介孔吸附材料,其氧化态、新鲜还原态、反应后的状态之间存在结构变化。5种催化剂的表面碱性吸附位在数量上均以弱碱位为辅,以强碱位为主。
(2)Fe-K2O/MAl2O4催化剂的费-托合成反应活性由高到低的次序为Fe-K2O/Sr Al2O4、Fe-K2O/BaAl2O4、Fe-K2O/CaAl2O4、Fe-K2O/MgAl2O4、Fe-K2O/Mn Al2O4。在310℃时,5种催化剂的C2~C4烯/烷比由高到低的次序为Fe-K2O/Mn Al2O4、Fe-K2O/MgAl2O4、Fe-K2O/BaAl2O4、Fe-K2O/CaAl2O4、Fe-K2O/Sr Al2O4。5种催化剂的抑制加氢能力与其表面碱性位的碱性强度大致呈反向关系。5种催化剂的C-C偶合能力、低碳烯烃选择性亦均与其表面碱性位的碱性强度大致呈反向关系。催化剂表面碱性位的碱性强度对催化剂的活性有非常重要的影响,即表面碱性位的碱性越强则催化剂的活性越高。催化剂表面碱性位的碱性越强则越容易被反应温度的升高引发二次加氢反应。
(3)5种催化剂的C2=~C4=烃类选择性最大值由高到低的次序为Fe-K2O/MgAl2O4、Fe-K2O/MnAl2O4、Fe-K2O/CaAl2O4、Fe-K2O/BaAl2O4、Fe-K2O/Sr Al2O4。为了提高产品中低碳烯烃的烃类选择性,催化剂表面碱性位的碱性强度要适度(即CO2-TPD高温脱附峰温度不宜高于700℃)。
