Pd掺杂Fe催化剂上邻甲酚加氢脱氧理论研究
2021-08-24张泽石聂小娃宋春山郭新闻
张泽石聂小娃宋春山郭新闻
(1.大连理工大学 化工学院 精细化工国家重点实验室PSU-DUT联合能源研究中心,辽宁 大连 116024;2.香港中文大学 理学院 化学系,中国香港 沙田 999077)
由于传统化石燃料的消耗和在使用过程中产生的环境问题,开发清洁能源和可再生能源变得尤为重要[1-3]。生物质是地球上最广泛存在的物质之一,具有可再生、污染少、成本低等特点。生物质作为一种可再生的碳资源而备受关注,其也是用于生产有价值化学品和燃料的一种重要资源[4-6]。木质素是生物质的重要组成部分。快速热解是一种直接将木质素基生物质转化为生物油的有效热化学方法。然而,木质素热解过程中能够产生大量的酚类化合物,对生物油的应用会产生负面影响,如降低热值和稳定性、影响挥发性和黏度等[7]。因此,这些生物油用作燃料之前要经过进一步加工脱氧[8-9]。酚类化合物转化为烃类产品是生物油加工升级的关键步骤。催化加氢脱氧(Hydrodeoxygenation,HDO)是一种理想的方法,可以选择性地将酚类化合物转化为有价值的碳氢化合物[10-13]。近年来,木质素基酚类化合物在温和反应条件下进行选择性加氢脱氧制备液体芳烃(苯(Benzen)、甲苯(Toluene)、二甲苯(Xylene),统称BTX)成为研究的重点[14]。BTX是有机化工的重要原料,可广泛应用于燃料添加剂、溶剂、涂料、树脂、染料等领域。通过调节催化剂的组成和结构,对酚类化合物进行选择加氢脱氧生成芳烃具有重要的应用前景和研究价值。
具有亲氧性的Fe催化剂因其对HDO反应具有独特的选择性和较低的成本,近年来受到越来越多的关注[15-16]。虽然Fe催化剂的活性不如贵金属,但它对酚类化合物CAr—O键的活化和断裂具有独特的选择性,有利于生成芳烃产物[16-17]。桑小义等[18]研究了邻甲酚在还原态Mo/Al2O3、Co Mo/Al2O3和Co MoEDTA/Al2O3催化剂上加氢脱氧的转化率及产物的选择性差异,采用还原态Mo/Al2O3催化邻甲酚加氢脱氧时产物以直接脱氧产物甲苯为主。Nie等[19]研究了在0.1 MPa和温度300℃下,SiO2负载单金属Ni、Fe及双金属Fe-Ni催化剂上间甲酚的HDO反应。结果表明:在单金属Ni催化剂上,以3-甲基环己醇为主要产物;而在单金属Fe和双金属Fe-Ni催化剂上,以甲苯为主要产物,证明Fe基催化剂能够抑制苯环加氢生成环饱和产物。Hong等[20]研究发现,Fe-Pd双金属催化剂能够选择性地促进间甲酚CAr—O键断裂,在不发生苯环加氢和C—C键氢解的情况下高选择性生成甲苯,得到的产物分布与单金属Fe催化剂相近,但HDO活性在加入Pd之后得到较大提升。Han等[21]研究了Fe-Ni双金属催化剂上苯酚的HDO反应,指出金属Ni位点和Ni-Fe合金活性位点之间的协同作用促进H2分子活化和CAr—O键的裂解,有利于提高苯酚的HDO反应性能。Fe-Ni催化剂上的吸附活性位点比单金属Ni上的吸附活性位点多,显著提高了催化活性。这些结果为优化设计HDO催化剂的新策略提供了研究基础和指导。在Fe催化剂中掺杂Ni、Pd等过渡金属有利于H2分子的活化和解离,提高表面H*物种的覆盖度,进而影响反应分子和关键中间体的表面吸附强度,最终加快HDO反应速率。Sirous-Rezaei等[22]研究了不同加氢金属和弱酸性载体对间甲酚和愈创木酚在温和反应条件下(温度小于350℃,压力0.1 MPa)转化成芳烃的HDO活性影响。其中,Pd-ReOx/Zr O2催化剂表现出最高的HDO活性,得到甲苯的产率为77.2%(质量分数);金属和载体的HDO催化活性由大到小顺序依次为Pd、Ni、Ru、Cu和Zr O2、CeO2、Zr CeO2,在这些催化剂上均没有环加氢产物生成;Pd、Ru、Cu金属催化剂上有利于间甲酚选择性转化为甲苯;Ni催化剂促进间甲酚脱甲基反应,除甲苯外还生成了苯酚进而转化成大量的苯产物。Kwon等[23]通过X射线光电子能谱(XPS)、扫描透射电子显微镜(STEM)、X射线吸收精细结构光谱(XANES)表征,发现Pd原子分散在被还原的Fe纳米颗粒表面,产生了较强的Pd-Fe相互作用;结合密度泛函理论(DFT)计算揭示了Pd在Fe催化剂中的作用,Pd的加入促进了H2的解离,生成表面H*物种,提高了金属Fe表面的稳定性并且产物的解吸也得到了增强。因此,在Fe-Pd双金属催化剂上,HDO反应活性和芳烃选择性均得到提高。然而,影响这些Fe基双金属催化剂性能和HDO反应途径的重要因素并没有在这些研究中进行深入讨论。
虽然Fe基双金属催化剂对酚类化合物(如苯酚、间甲酚)加氢脱氧提高芳烃选择性具有潜在的优势,但其活性中心的结构性质、分子反应机理、双金属协同催化的微观本质以及催化剂构-效关系等方面仍然是有待阐明的重要科学问题。对于不同酚反应物分子,其加氢脱氧性能对催化剂组成和结构有何依赖?过渡金属掺杂Fe催化剂的微观作用机制是怎样的?加氢脱氧反应不可避免水的生成,其对反应性能将产生怎样的影响?这些重要问题仍需要进一步深入探讨。
笔者选取邻甲酚作为木质素基酚类化合物的模型反应物,选取带有阶梯(Step)的Fe(211)表面作为Fe催化剂模型,利用密度泛函理论方法研究了邻甲酚加氢脱氧反应机理和催化性能。在此基础上,进一步探究在Fe催化剂中掺杂Pd对HDO活性和产物选择性的影响。此外,还研究了表面生成的H2O*对邻甲酚加氢脱氧反应性能的影响,考虑了不同的H2O*作用形式。此研究为理解Fe基双金属催化酚类化合物加氢脱氧反应机理和影响催化性能的重要因素提供了理论基础,揭示了Pd掺杂及水参与对Fe催化剂活性及产物选择性的潜在影响,为未来高效催化剂的优化设计提供了重要思路。
1 计算方法
采用维也纳大学Hafner小组开发的VASP(Vienna ab-initio simulation package)软件包,在密度泛函理论(DFT)框架内进行自旋极化计算[24]。采用投影缀加平面波(PAW)赝势处理电子-离子相互作用[25]。交换和相关能的计算采用广义梯度近似(GGA)方法和Perdew,Burke和Ernzerhof(PBE)泛函[26]。平面波基组的截断能设置为400 e V。使用VASP程序中的阻尼分子动力学方法进行结构优化,直到所有原子运动单位距离上的能量都小于0.2 eV/nm作为收敛标准。过渡态优化采用爬坡弹性带(The climbing image nudged elastic band,CI-NEB)方法进行计算,并验证每个过渡态沿反应坐标方向仅有一个虚振动频率[27]。采用Henkelman课题组开发的巴德电荷(Bader charge)计算方法进行电荷转移情况分析[28]。
体相Fe采用体心立方(bcc)结构,基于实验测定的晶格常数(0.2866 nm),构建了Fe催化剂的阶梯(211)晶面。包括4个原子层,其中底部两层在结构优化过程中被固定,而顶部两层以及吸附分子则完全放开进行结构优化。所有表面计算都采用Monkhorst-Pack方案中的(2×2×1)k点进行,并设置1.2 nm的真空层来避免重复单元之间的相互作用。在Fe(211)表面引入1个Pd原子来取代1个顶层Fe原子,从而构建出Pd-Fe(211)表面模型,用于研究Pd掺杂的作用和影响。
表面物种的吸附能(Eads,e V)由公式Eads=EA-S-ES-EA计算,其中EA-S为催化剂表面与吸附物种相互作用的总能量(eV),ES为空表面的能量(e V),EA为吸附物种在气相中的能量(e V)。Eads越负,表明催化剂表面与吸附物种之间的相互作用越强。基元反应的活化能垒(Eact,e V)根据公式Eact=ETS-EIS计算,其中ETS为基元反应过渡态的能量(e V),EIS为指定基元反应步骤中反应物吸附态的能量(e V)。基元反应中的反应能(Erxn,eV)由公式Erxn=EFS-EIS计算,其中EFS为生成产物吸附在催化剂表面上的能量(eV)。根据计算得到的吸附能(Eads)、反应能(Erxn)和活化能垒(Eact),绘制出邻甲酚在纯Fe(211)及Pd掺杂Fe(211)表面上进行HDO反应的能量路径。根据阿伦尼乌斯方程(k=Ae-Ea/RT)估算特定反应温度下生成产物的相对反应速率差异。例如:2种产物的活化能差异为0.1 e V,反应温度为300℃,那么二者的相对反应速率约为7.5;如果反应温度升高到400℃,那么二者的相对反应速率约为31,因此生成产物的相对反应速率差异大小与反应温度相关。
2 邻甲酚在Fe(211)表面上的吸附和转化路径
邻甲酚的吸附和活化是其进一步转化的关键前期步骤。苯环中H—Π键、CAr—C键以及CAr—O键的活化方式和程度直接影响反应路径和产物选择性。邻甲酚的吸附构型和吸附强度对HDO反应性能具有一定影响。首先考察了邻甲酚在Fe(211)表面上的吸附构型,计算了吸附能和结构参数。
2.1 邻甲酚在Fe(211)表面上的吸附位点和吸附构型
Fe(211)是一种阶梯式表面,包含六重中空位点(6-Fold hollow site)、顶点(Top site)和桥位点(Bridge site)。总体考虑了两类吸附构型,即垂直吸附(A)和水平吸附(B)。垂直吸附是指依靠邻甲酚的—OH基团与Fe(211)表面直接相互作用;水平吸附是指依靠邻甲酚苯环的Π键与Fe(211)表面发生相互作用。在Fe(211)表面上得到了4种稳定的吸附构型,如图1所示。各吸附构型的吸附能和关键结构参数见表1。在垂直吸附构型中(见图1(a)),邻甲酚的—OH基团与Fe(211)表面发生吸附作用,但相互作用不强,吸附能仅为-0.38 eV。在水平吸附模式下(见图1(b)~(d)),苯环趋于平行Fe(211)表面,平行吸附构型与Fe表面的相互作用要比垂直吸附构型强很多(见表1吸附能数据)。由于Fe(211)表面和H原子之间的排斥力,苯环碳氢键略微向上倾斜。计算得到的邻甲酚最稳定吸附构型为B-s1(见图1(b)),吸附能为-0.91 e V,苯环与Fe(211)表面的平均距离为0.191 nm。平行吸附构型可能引起苯环H—Π键、CAr—C键和CAr—O键的活化,其活化程度直接影响邻甲酚转化路径和产物的选择性;垂直吸附构型可能有利于CAr—O键的活化和断裂,具体计算结果将在下节中进行讨论。
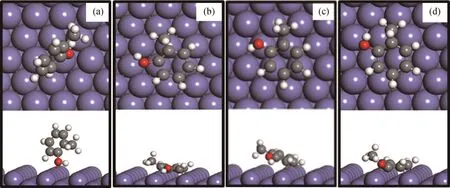
图1 邻甲酚在Fe(211)表面上的吸附构型Fig.1 Adsorption configurations of o-cresol molecule on Fe(211)surface
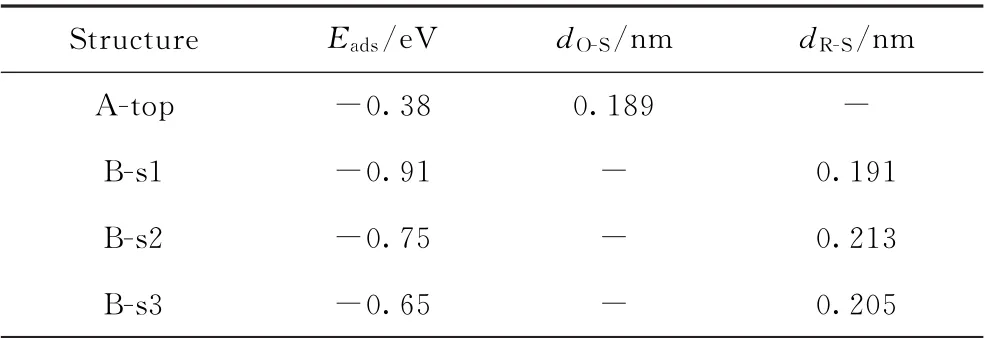
表1 邻甲酚在Fe(211)表面上的吸附能(E ads)和关键结构参数Table 1 Adsorption energies(E ads)and key structural parameters for o-cresol adsorption on Fe(211)surface
2.2 邻甲酚在Fe(211)表面上的加氢脱氧反应机理
CAr—C键和CAr—O键的活化方式和程度决定了邻甲酚通过直接脱羟基再加氢生成甲苯,或者是脱甲基再加氢生成苯酚的不同转化路径。此研究中系统地探讨了邻甲酚在Fe(211)表面上的加氢脱氧反应机理,计算了2种途径的反应能和活化能垒,绘制出反应能线图。为了实现高选择性生产芳烃,催化剂应具有较好的对邻甲酚CAr—O键的活化和断裂活性。
2.2.1 生成苯酚的反应路径
首先,基于邻甲酚通过苯环在Fe(211)表面的最稳定吸附构型(水平吸附,将图1(b))以及通过—OH基团吸附(垂直吸附,见图1(a)),研究了邻甲酚脱甲基再加氢生成苯酚的反应途径。在考察基元反应步骤(C7H7OH*→C6H4OH*+CH3*和C6H4OH*+H*→C6H5OH*)时,水平吸附的邻甲酚的CAr—C键被活化并断裂,从而形成C6H4OH*中间体和CH3*物种吸附在Fe(211)表面上;然后,1个氢原子(H*)被放置在Fe(211)表面取代生成的CH3*物种。H*与C6H4OH*中间体结合导致表面上形成苯酚分子。图2为最稳定吸附的邻甲酚在Fe(211)表面上脱甲基生成C6H4OH*再加氢生成苯酚的反应初态(IS)、过渡态(TS)、终态(FS)的最优结构。可以看出:水平吸附的邻甲酚(见图2(a)-IS)的CAr—C键断裂能够在表面阶梯位点形成吸附的CH3*物种,而C6H4OH*中间体则从最初吸附位置远离CH3*物种(见图2(a)-FS)。过渡态的CAr—C键距离为0.205 nm(见图2(a)-TS),并且沿着CAr—C断裂方向具有1个452.94 cm-1的虚振动频率。邻甲酚脱甲基反应的能垒为1.40 e V,反应能为-0.50 eV。C6H4OH*中间体继续加氢生成苯酚需要克服的反应能垒为0.69 e V,反应能为-0.13 eV。当邻甲酚通过—OH基团与Fe(211)表面发生吸附作用时,由于CAr—CH3*物种距离表面较远,因此CAr—C键不能够被有效活化,邻甲酚脱甲基的反应能垒高达1.87 e V,形成了极不稳定的过渡态结构,因此在动力学上不具有优势。
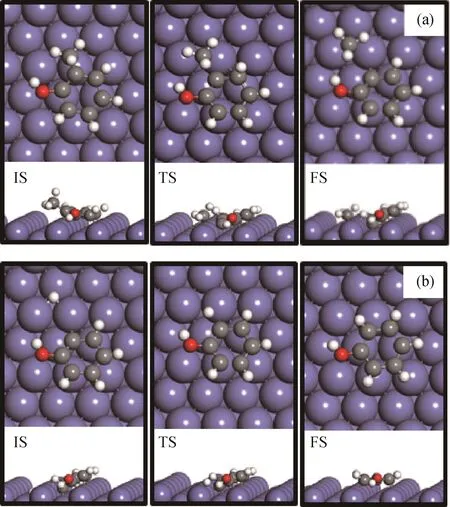
图2 最稳定吸附的邻甲酚在Fe(211)表面上脱甲基再加氢生成苯酚的反应初态(IS)、过渡态(TS)、终态(FS)的最优结构Fig.2 Optimized configurations of initial(IS),transition(TS)and final(FS)states associated with direct demethylation pathway of o-cresol conversion to phenol started with the most stable horizontal adsorption configuration on Fe(211)surface
2.2.2 生成甲苯的反应路径
为了高选择性生产芳烃产物,要求催化剂对CAr—O键活化断裂具有较高的活性。同样基于邻甲酚通过苯环在Fe(211)表面的最稳定吸附构型(水平吸附,见图1(b))以及通过—OH基团吸附(垂直吸附,见图1(a))2种不同的吸附构型,研究了邻甲酚通过直接脱羟基再加氢生成甲苯的反应途径。在考察基元反应步骤(C7H7OH*→C7H7*+OH*和C7H7*+H*→C7H8*)时,水平吸附的邻甲酚的CAr—O键被活化并断裂,从而形成C7H7*中间体和OH*物种吸附在Fe(211)催化剂表面上;然后,用1个氢原子(H*)取代表面生成的OH*物种。H*与C7H7*中间体结合导致表面形成甲苯分子。图3为最稳定吸附的邻甲酚在Fe(211)表面上直接脱羟基生成C7H7*再加氢生成甲苯反应初态(IS)、过渡态(TS)、终态(FS)的最优结构。可以看出:水平吸附的邻甲酚(见图3(a)-IS)的CAr—O键断裂能够在表面阶梯位点形成吸附的OH*物种,而C7H7*中间体则从最初吸附位置远离OH*(见图3(a)-FS)。由于分子不饱和,形成的C7H7*中间体表现出明显的结构变形(图3(a)中FS与IS结构相比)。过渡态的CAr—O键距离为0.200 nm(见图3(a)-TS),并且沿着CAr—O键断裂方向存在1个320.75 cm-1的虚振动频率。邻甲酚脱羟基反应能垒为1.01 e V,反应能为-0.58 e V。C7H7*中间体加氢继续生成甲苯需要克服反应能垒为0.43 e V,反应能为-0.25 e V。当邻甲酚通过—OH基团与Fe(211)表面发生吸附作用时,虽然CAr—OH*物种距离表面较近,但是脱羟基过程产生的C7H7*中间体不能够被Fe(211)表面所稳定,导致邻甲酚直接脱羟基的反应能垒高达1.59 e V,与水平吸附构型相比,此脱羟基反应在动力学上是不利的。计算结果表明,邻甲酚通过—OH基团吸附于Fe(211)表面并没有促进CAr—O键的活化和断裂,这是由于脱羟基过程中形成了极不稳定的过渡态结构。
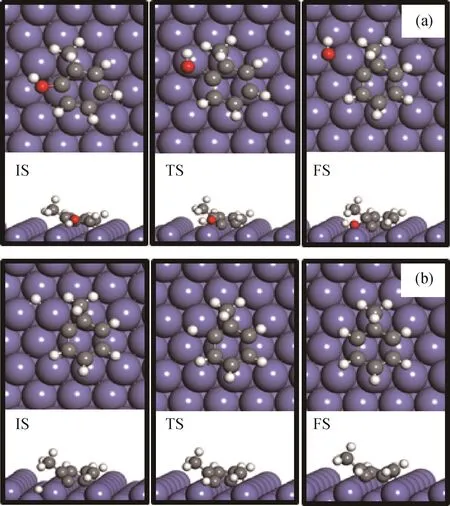
图3 最稳定吸附的邻甲酚在Fe(211)表面上直接脱羟基再加氢生成甲苯反应初态(IS)、过渡态(TS)、终态(FS)的最优结构Fig.3 Optimized configurations of initial(IS),transition(TS)and final(FS)states associated with the direct dehydroxylation pathway of o-cresol conversion to toluene started with the most stable horizontal adsorption configuration on Fe(211)surface
2.2.3 邻甲酚在Fe(211)表面上加氢脱氧反应的产物选择性
为了比较邻甲酚在Fe(211)表面上直接脱羟基再加氢生成甲苯以及脱甲基再加氢生成苯酚的产物相对选择性,采用DFT方法计算了2种路径各基元反应的活化能垒,估算了生成2种产物的反应速率常数之比。表2为所涉及的各个基元步骤的反应能、活化能垒以及过渡态的关键结构参数。DFT计算结果表明,邻甲酚在Fe(211)上直接脱羟基反应相比于脱甲基反应能垒低0.39 eV,之后的加氢反应能垒依然低0.26 e V。因此,纯Fe(211)表面对邻甲酚脱羟基反应具有更高的催化活性,有利于产物甲苯的生成。图4为邻甲酚经脱羟基和脱甲基以及后续加氢生成产物甲苯和苯酚的反应能线图。比较2种路径总反应的活化能垒,邻甲酚在Fe(211)表面上转化生成甲苯总反应的活化能垒为1.01 eV,而经脱甲基生成苯酚总反应的活化能垒为1.40 e V,表明邻甲酚加氢脱氧转化为甲苯更具动力学优势。基于甲基酚化合物加氢脱氧的相关实验研究,选取反应温度为350℃,根据阿伦尼乌斯方程估算得到生成甲苯和苯酚的相对选择性约为1.4×103。Hong等[29]在300℃下将H2和反应物气体混合通入固定床反应器测试了不同H2、H2O及间甲酚压力下,间甲酚加氢脱氧生成甲苯的选择性均在91%以上,且基于CAr—O直接断裂机制的动力学模型可以很好地描述Fe催化剂上间甲酚HDO反应的动力学行为。
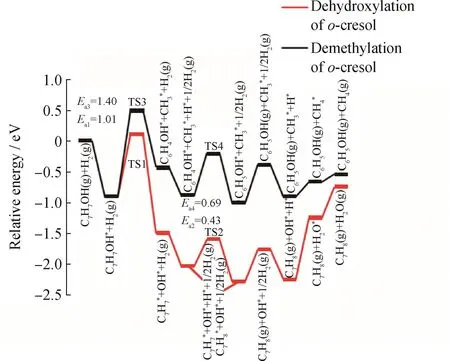
图4 Fe(211)上邻甲酚直接脱羟基再加氢生成甲苯以及脱甲基再加氢生成苯酚的反应能线图Fig.4 Energy diagrams for direct dehydroxylation of o-cresol followed by hydrogenation to produce toluene and demethylation of o-cresol followed by hydrogenation to produce phenol on Fe(211)surface
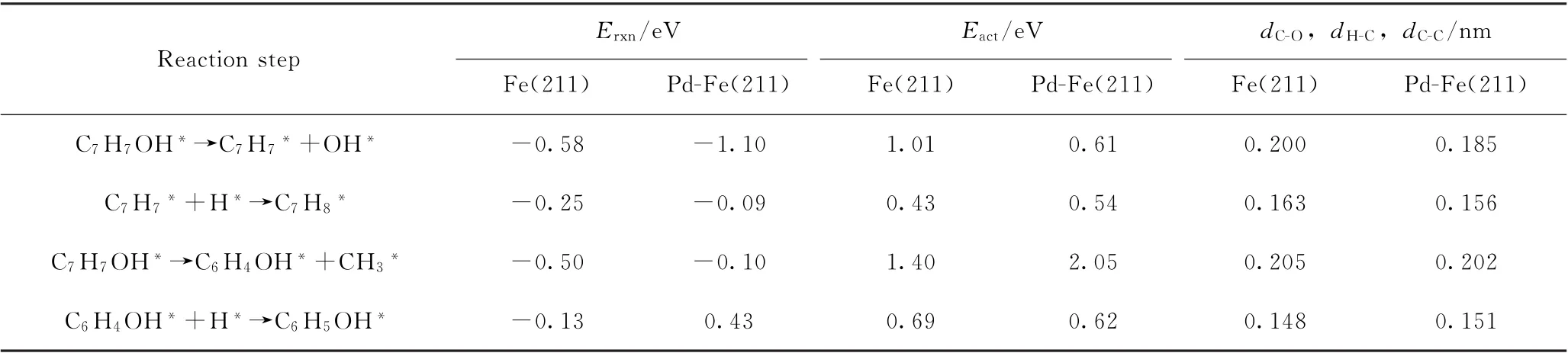
表2 Fe(211)及Pd-Fe(211)表面上邻甲酚直接脱羟基再加氢生成甲苯以及脱甲基再加氢生成苯酚相关的反应能(E rxn)、活化能垒(E act)和关键结构参数Table 2 Reaction energies(E rxn),activation barriers(E act)and key structural parameters of transition states associated with direct dehydroxylation of o-cresol to produce toluene and demethylation to produce phenol on Fe(211)and Pd-Fe(211)surfaces
3 邻甲酚在Pd-Fe(211)表面上的吸附和转化路径
在Pd-Fe(211)表面上,首先考察了邻甲酚的吸附构型,计算了吸附能和结构参数。探究掺杂Pd对邻甲酚吸附结构和稳定性的影响,并选取最稳定吸附构型及Pd与邻甲酚羟基不同相对位置上的吸附构型,研究其在Pd-Fe(211)表面上进行加氢脱氧反应生成苯酚和甲苯的反应机理,揭示掺杂Pd对催化活性和产物选择性的影响。
3.1 掺杂Pd对邻甲酚吸附结构和稳定性的影响
同样考虑了邻甲酚在Pd-Fe(211)表面的两类吸附构型,即垂直吸附和水平吸附。在Pd-Fe(211)表面上得到了5种稳定的邻甲酚吸附结构,如图5所示。表3为邻甲酚在Pd-Fe(211)表面的吸附能和关键结构参数。
由图5(a)、(b)可见,在垂直吸附构型中,邻甲酚同样以—OH基团与Pd-Fe(211)表面发生吸附作用,但相互作用不强,吸附能分别为-0.36和-0.39 eV。相比于Fe(211)表面上的垂直吸附构型(见图1(a),Eads=-0.38 eV),吸附能差异不大。由图5(c)~(e)可见,在水平吸附模式下,苯环平行于Pd-Fe(211)表面,平行吸附构型与表面相互作用比垂直吸附构型强很多。由于Fe表面和氢原子之间的排斥力,C—H键略微向上倾斜(见图5(e))。计算出邻甲酚最稳定的吸附构型为B-s1(见图5(c)),吸附能为-0.70 eV,苯环与催化剂表面的平均距离为0.230 nm。此时,邻甲酚的羟基基团距离掺杂Pd原子较近。比较3种水平吸附构型、结构参数和吸附能,发现邻甲酚的苯环与Fe表面的相对位置以及—OH基团与掺杂Pd原子的相对位置对邻甲酚分子的吸附构型和稳定性具有一定影响(见表3)。与Fe(211)相比,掺杂Pd减弱了邻甲酚的吸附强度,吸附能降低0.21 e V,且苯环相对于表面的距离变大。
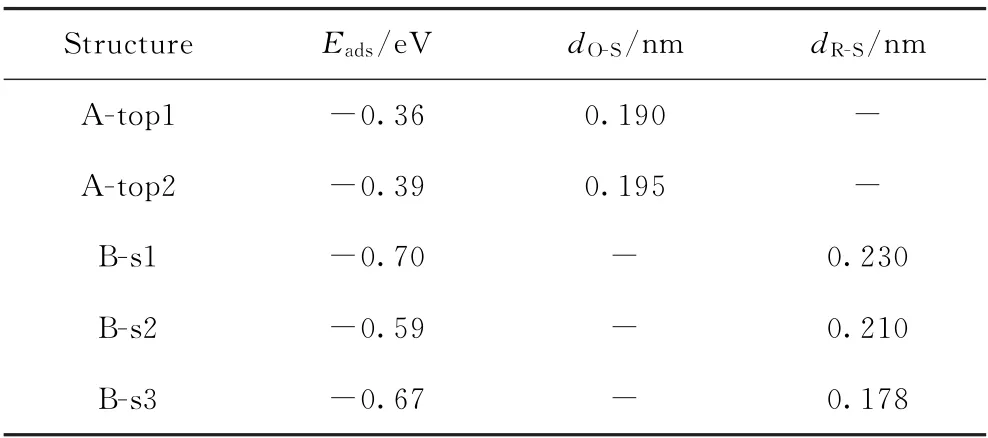
表3 邻甲酚在Pd-Fe(211)表面的吸附能(E ads)和关键结构参数Table 3 Adsorption energies(E ads)and key structure parameters for o-cresol adsorption on Pd-Fe(211)surface
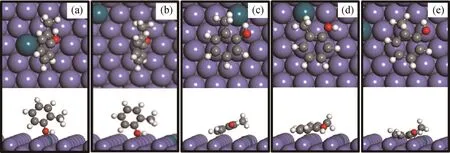
图5 邻甲酚在Pd-Fe(211)表面上的吸附构型Fig.5 Adsorption configurations of o-cresol molecule on Pd-Fe(211)surface
3.2 邻甲酚在Pd-Fe(211)表面上加氢脱氧反应机理
上述DFT计算结果表明,Fe催化剂中掺杂Pd对邻甲酚的吸附性能具有一定影响,进而可能会影响反应分子的活化性能和加氢脱氧反应路径。Fe(211)和Pd-Fe(211)表面都有利于邻甲酚通过苯环进行吸附活化,而CAr—C键和CAr—O键的活化程度决定了邻甲酚通过直接脱甲基或者脱羟基不同途径进行转化。在此研究中,系统地探讨了邻甲酚在Pd-Fe(211)表面上的加氢脱氧机理,计算了2种反应路径的反应能量和活化能垒,绘制了反应能线图。为了高选择性生产芳烃,催化剂应具有较高的对邻甲酚的CAr—O键活化和断裂活性。
3.2.1 生成苯酚的反应路径
首先,基于邻甲酚通过苯环在Pd-Fe(211)表面上的最稳定吸附构型(见图5(c),B-s1),研究了邻甲酚通过脱甲基再加氢生成苯酚的反应途径,得到的最优结构如图6(a)所示。水平吸附的邻甲酚的CAr—CH3键断裂导致在表面阶梯位点形成吸附态C物种。脱甲基过渡态的CAr—CH3键距离为0.187 nm,脱甲基活化能垒为2.26 e V,反应能为-0.15 eV。生成的C6H4OH*中间体加氢继续生成苯酚需要克服活化能垒为0.84 e V,反应能为-0.25 eV。此外,还探究了掺杂Pd原子与邻甲酚—OH基团的相对位置对邻甲酚脱甲基转化为苯酚的影响。邻甲酚在Pd-Fe(211)表面上最稳定吸附构型的羟基基团距离Pd原子较近。因此,又基于邻甲酚羟基基团距离Pd原子较远的吸附构型(见图5(e),B-s3),研究了邻甲酚脱甲基再加氢生成苯酚的反应途径,得到的最优结构如图6(b)所示。脱甲基过渡态的CAr—CH3键距离为0.202 nm。脱甲基活化能垒为2.05 eV,反应能为-0.10 eV。生成的C6H4OH*中间体加氢生成苯酚需要克服活化能垒为0.62 eV,反应能为0.43 eV。由此可见,邻甲酚的羟基基团与掺杂Pd原子的相对位置不仅影响其吸附稳定性,对其进一步发生HDO反应也具有一定影响。
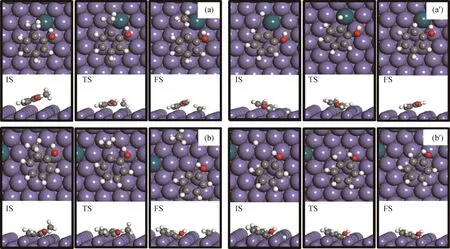
图6 邻甲酚在Pd-Fe(211)表面上直接脱甲基再加氢生成苯酚的反应初态(IS)、过渡态(TS)、终态(FS)的最优结构Fig.6 Optimized configurations of initial(IS),transition(TS)and final(FS)states associated with direct demethylation pathway of o-cresol conversion to phenol started with different adsorption configurations of o-cresol on Pd-Fe(211)surface
3.2.2 生成甲苯的反应路径
基于邻甲酚通过苯环在Pd-Fe(211)表面上的最稳定吸附构型(见图5(c),B-s1),研究了邻甲酚通过直接脱羟基再加氢生成甲苯的反应途径,得到的最优结构如图7(a)所示。水平吸附的邻甲酚的CAr—O键断裂导致在表面阶梯位点形成吸附态OH*物种,而剩下的C7H7*中间体在催化剂表面的相对位置变化不大。脱羟基过渡态的CAr—O键距离为0.198 nm,沿着C—O断裂方向具有1个392.82 cm-1的虚振动频率。脱羟基活化能垒为1.35 eV,反应能为-0.50 e V。形成的C7H7*中间体继续加氢生成甲苯需要克服活化能垒为0.40 eV,反应能为-0.31 e V。此外,还探究了掺杂Pd原子与邻甲酚—OH基团的相对位置对邻甲酚脱羟基转化为甲苯的影响。选取邻甲酚的羟基距离Pd原子较远的吸附构型(见图5(e),B-s3)作为初始态,计算了脱羟基反应路径,最优结构如图7(b)所示。脱羟基活化能垒仅为0.61 eV,反应能为-1.10 eV,在动力学和热力学上均具有明显优势。过渡态的CAr—O键距离为0.185 nm,其沿着C—O键断裂方向具有1个444.51 cm-1的虚振动频率。形成的C7H7*中间体继续加氢生成甲苯需要克服活化能垒为0.54 eV,反应能为-0.09 eV。由此可见,邻甲酚的羟基基团与掺杂Pd原子的相对位置对邻甲酚加氢脱氧性能具有重要影响,—OH基团远离Pd原子更有利于CAr—O键在Fe表面进行活化断裂,脱羟基能垒得到显著降低。

图7 邻甲酚在Pd-Fe(211)表面上直接脱羟基再加氢生成甲苯的反应初态(IS)、过渡态(TS)、终态(FS)的最优结构Fig.7 Optimized configurations of initial(IS),transition(TS)and final(FS)states associated with direct dehydroxylation of o-cresol followed by hydrogenation to produce toluene started with different adsorption configurations of o-cresol on Pd-Fe(211)surface
3.2.3 掺杂Pd对产物选择性的影响
邻甲酚在Pd-Fe(211)表面上直接脱羟基再加氢生成甲苯,以及脱甲基再加氢生成苯酚的各基元步骤的反应能、活化能垒以及过渡态的关键结构参数如表2所示。DFT计算结果表明,当邻甲酚的羟基基团远离掺杂Pd原子时,更有利于邻甲酚的CAr—O键或CAr—C键在Fe表面进行活化断裂;而当二者距离较近时,相应的CAr—O键或CAr—C键断裂能垒均升高。Pd掺杂对邻甲酚直接脱羟基再加氢生成甲苯表现出较高的催化活性,能垒显著降低(<0.7 eV);而CAr—C键断裂能垒较高(>2.0 eV),导致苯酚的生成在动力学上受阻。与Fe(211)相比,邻甲酚在Pd-Fe(211)上脱羟基的活化能垒从1.01 eV降低到0.61 e V,体现出Pd掺杂对甲苯生成的显著优势。图8为Fe(211)及Pd-Fe(211)表面上邻甲酚直接脱羟基再加氢生成甲苯的反应能线图。计算得到Fe(211)表面上邻甲酚加氢脱氧生成甲苯的总反应活化能垒为1.01 e V,而Pd-Fe(211)上则为0.61 e V,表明Pd的掺杂极大提高了甲苯生成反应速率(约提高1.6×103倍)。因此,在Fe(211)表面掺杂Pd有利于提高目标产物甲苯的选择性。
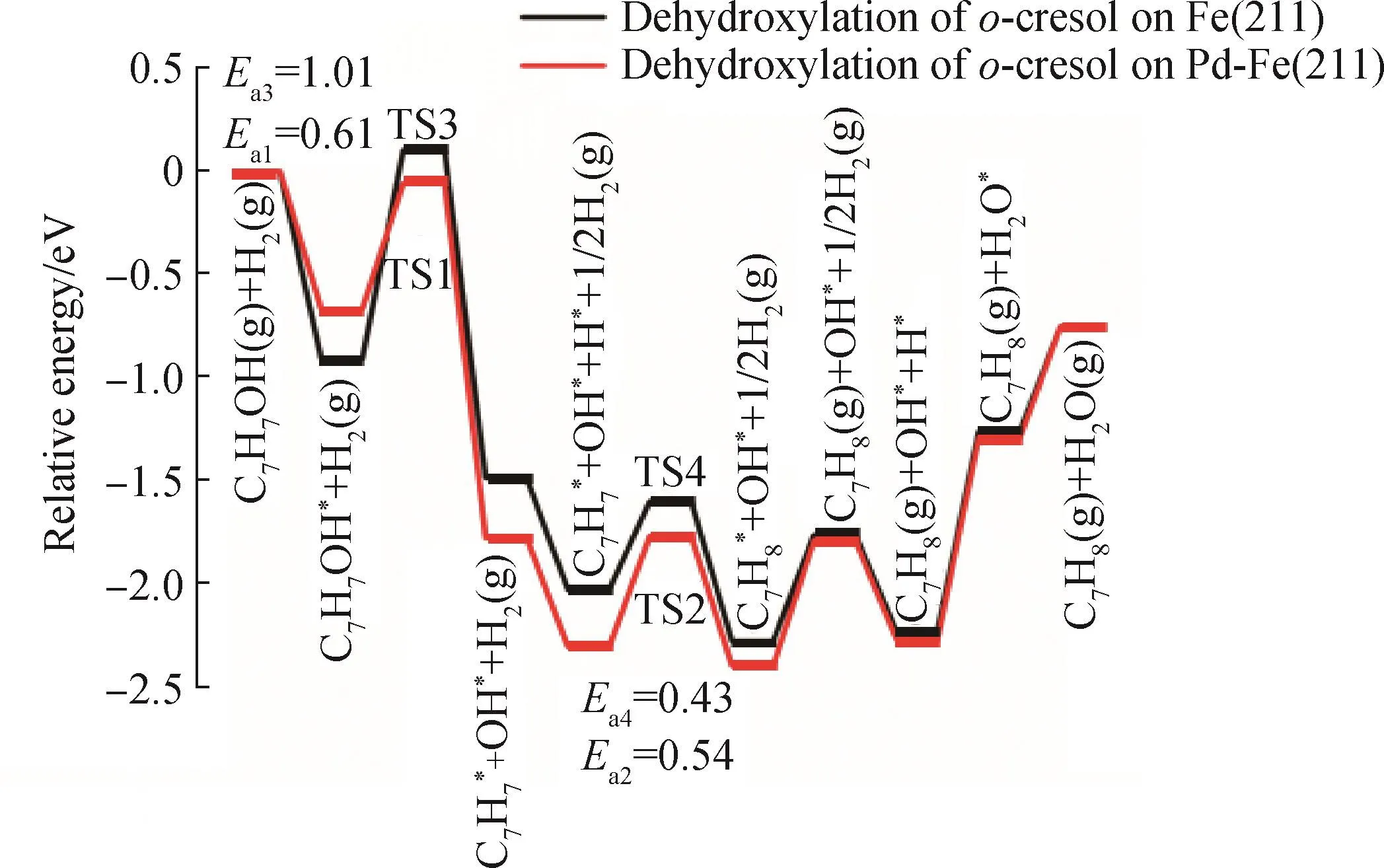
图8 Fe(211)及Pd-Fe(211)表面上邻甲酚直接脱羟基再加氢生成甲苯的反应能线图Fig.8 Energy diagrams for direct dehydroxylation of o-cresol followed by hydrogenation to produce toluene on Fe(211)and Pd-Fe(211)surfaces
从催化剂表面与吸附分子之间的电子转移状况分析了Fe(211)与Pd-Fe(211)产生不同催化活性的重要因素。图9为邻甲酚在Fe(211)和Pd-Fe(211)表面上直接脱羟基生成C7H7*中间体过程中形成过渡态结构的差分电荷密度图。可以看出,在Fe(211)上电子主要从Fe表面转移到邻甲酚的—OH上,表面与苯环之间也有少量电子转移,但表面与吸附物种之间的相互作用较弱(见图9(a))。而对于掺杂Pd之后的Pd-Fe(211)表面,同样处于最稳定吸附位(邻甲酚—OH靠近Pd原子),过渡态的结构显示电子主要从催化剂表面向邻甲酚的甲基上转移,羟基上的电子密度累积较小,表面与吸附物种之间的相互作用也不强(见图9(c))。而对于—OH基团距离掺杂Pd原子较远吸附位置下邻甲酚脱羟基形成的过渡态,可以看到大量电子从催化剂表面向邻甲酚的苯环及—OH基团转移,表面与吸附物种的相互作用得到显著增强(见图9(b)),使过渡态结构更加稳定。因此,Pd掺杂Fe催化剂提高了邻甲酚的加氢脱氧活性和产物甲苯的选择性,电子转移分析结果与反应能垒计算趋势相一致。
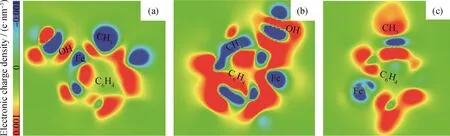
图9 邻甲酚在Fe(211)及Pd-Fe(211)表面上直接脱羟基生成C7 H7*过渡态结构的差分电荷密度图Fig.9 Electronic charge density difference of the transition state structures associated with o-cresol dehydroxylation to form the C7 H7*intermediate on Fe(211)and Pd-Fe(211)surfaces
此外,还计算了Fe(211)和Pd-Fe(211)表面上邻甲酚加氢脱氧2种反应路径中第一步脱羟基和脱甲基初态及过渡态的电荷转移情况,并与反应能垒结果进行关联。表4为邻甲酚的CAr—O键和CAr—C键断裂的2种反应路径的初态(IS)、过渡态(TS)的反应分子与催化剂表面的电荷转移量及活化能垒。通过比较2种反应路径从初态到过渡态电荷转移量的差异,进而说明过渡态的稳定性和反应性能。从表4可以看出,除Pd-Fe(211)表面上的脱甲基反应外,其他3种情况形成的过渡态相对于初态反应分子上的电荷转移量均增加。Pd-Fe(211)表面上初态的邻甲酚分子与催化剂表面电荷转移量多于Fe(211)表面,说明邻甲酚分子在Pd掺杂的Fe(211)上活化程度较大;而且Pd-Fe(211)表面上脱羟基反应形成的过渡态分子上的总负电荷最多,意味着过渡态分子与催化剂表面的电子相互作用更强,这种强相互作用增加了过渡态的稳定性,从而降低了直接脱羟基反应的活化能垒。而对于Pd-Fe(211)表面上邻甲酚脱甲基,所形成的过渡态上总负电荷最少,从初态到过渡态的电荷转移量变化也最小,表明催化剂表面与过渡态的相互作用较弱,过渡态不稳定,导致反应能垒升高。这些电荷性质分析结果与反应能垒的计算趋势相吻合。
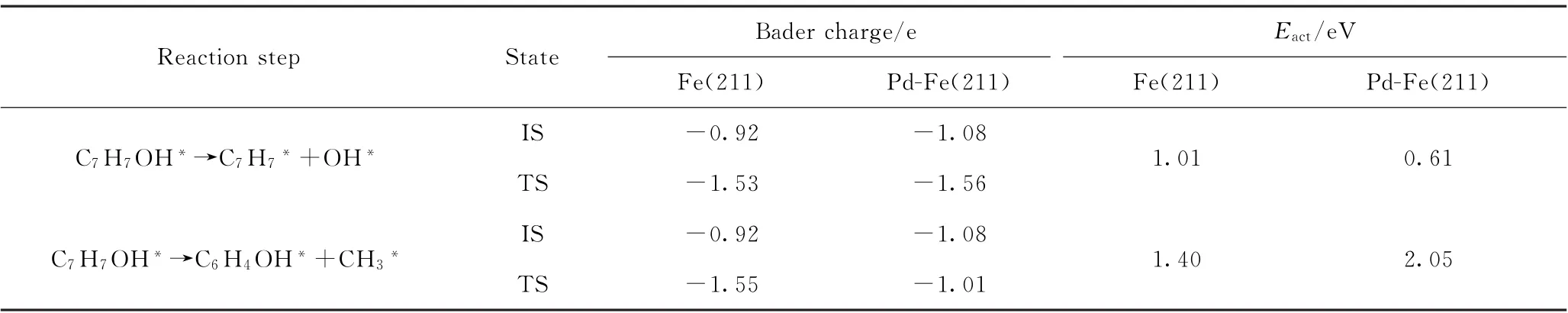
表4 邻甲酚的CAr—O键和CAr—C键断裂2种反应路径的初态(IS)、过渡态(TS)的电荷转移及反应的活化能垒(E act)Table 4 Charge transfer of initial state(IS)and transition state(TS)associated with the two reaction pathways of CAr—O and CAr—C cleavage of o-cresol and activation barriers(E act)
3.2.4 掺杂Pd对H2分子活化解离的影响
计算了H2分子在Fe(211)及Pd-Fe(211)上的吸附和解离过程,结果表明,当将H2分子放置到2个催化剂表面时,其发生自发解离,形成2个吸附态H原子。在Fe(211)上H2的解离吸附能为-1.14 eV,在Pd-Fe(211)上H2的解离吸附能为-1.25 e V,表明掺杂金属Pd有利于H2的快速活化和解离,导致表面H*物种的覆盖度增加。
在此基础上,选取Pd-Fe(211)表面,计算了当表面存在1个吸附态H*和3个H*情况下邻甲酚的吸附能及其加氢脱氧生成甲苯的活化能垒和反应能,计算结果如表5和表6所示。随着表面H*覆盖度的增加,邻甲酚的吸附强度有所减弱(见表5),在Fe(211)和Pd-Fe(211)上表现出相同的规律,计算结果与苯酚在Fe-Pd双金属上加氢脱氧理论研究结果[30]相一致。对于H*表面覆盖度对邻甲酚加氢脱氧生成甲苯反应的影响,计算结果表明在Fe(211)表面上,邻甲酚直接脱羟基生成C7H7*中间体物种受表面H*覆盖度的影响较小,从0H*→1H*→3H*随表面覆盖度增加,反应的活化能垒变化小于0.12 e V(见表6)。此外,C7H7*中间体物种进一步加氢生成甲苯的活化能垒随着表面H*覆盖度增加变化不大(小于0.1 eV)。在Pd-Fe(211)表面上表现出相同的趋势(见表6)。由此看来,虽然表面H*覆盖度增加会降低邻甲酚在催化剂表面上的吸附能,但对其加氢脱氧生成甲苯的反应性能影响不大。Pd的引入能够加速H2分子的活化解离,为反应提供充足的活性H*,有利于提高加氢脱氧速率。
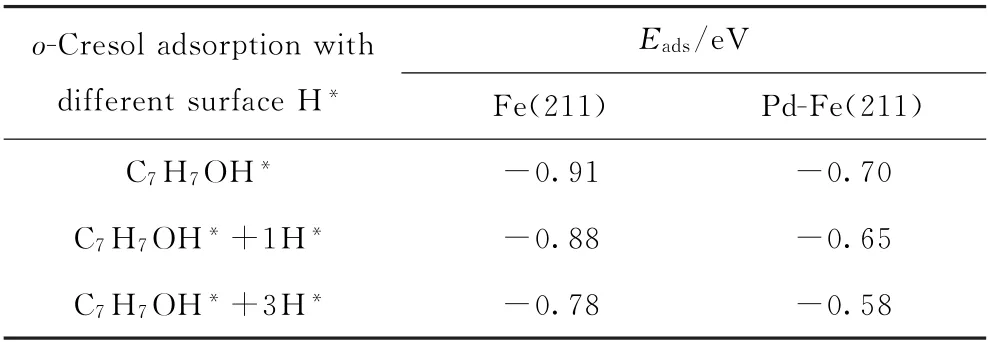
表5 在具有1H*和3H*的Fe(211)及Pd-Fe(211)表面上最稳定邻甲酚吸附构型的吸附能(E ads)Table 5 Adsorption energy(E ads)of the most stable o-cresol adsorption configuration on the surface of Fe(211)and Pd-Fe(211)in the presence of 1H*and 3H*
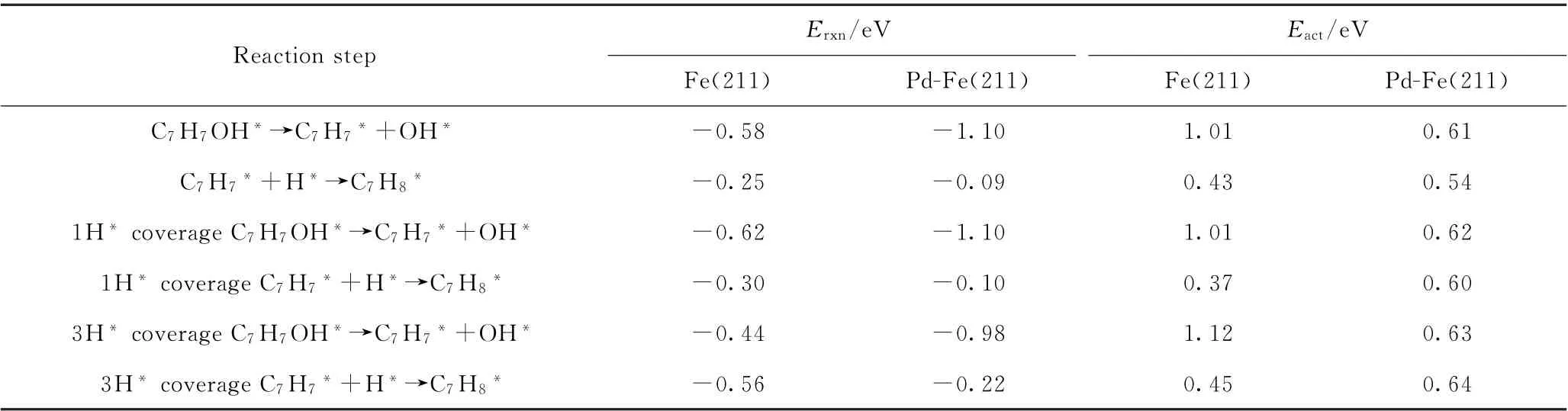
表6 在具有1H*和3H*的Fe(211)及Pd-Fe(211)表面上邻甲酚直接脱羟基再加氢生成甲苯的反应能(E rxn)和活化能垒(E act)Table 6 Reaction energies(E rxn)and activation barriers(E act)associated with direct dehydroxylation of o-cresol followed by hydrogenation to produce toluene on Fe(211)and Pd-Fe(211)surfaces in the presence of 1H*and 3H*
Hong等[29]构建的动力学模型以及XPS、STEM、XANES表征表明,贵金属Pd的掺杂可显著改变Fe催化剂上间甲酚的HDO反应速率常数。且Pd的加入有利于H2活化解离,提高表面H*的覆盖度,能够为Fe催化剂表面关键物种的加氢反应提供更多活性H*,有利于提高HDO速率。Pd辅助H2解离能够提高金属Fe催化剂表面的稳定性并加快产物解吸。上述DFT计算结果表明,Pd掺杂Fe(211)催化剂能够通过调节表面电子性质及表面-吸附物种间相互作用进而降低邻甲酚加氢脱氧生成甲苯的反应活化能,与Fe(211)相比,Pd-Fe(211)的甲苯选择性得到进一步提高。Pd-Fe双金属产生的协同催化作用可能同样适用于其他贵金属掺杂Fe催化剂体系,为未来高活性和高选择性HDO催化剂的优化设计提供了具有前景的策略和研究思路。
4 H2 O对邻甲酚加氢脱氧反应性能的影响
H2O*是酚类化合物加氢脱氧反应不可避免的副产物,OH*物种与活化H*反应生成水具有可逆性[31]。邻甲酚HDO生成甲苯等芳烃是预期的目标产物。此研究进一步探讨了表面H2O*物种对邻甲酚在Fe(211)及Pd-Fe(211)表面上进行脱羟基再加氢生成甲苯反应的影响。考虑了H2O*发生作用的2种不同机理,分别为氢键(H-bonding)机理和氢传递(H-shuttling)机理。氢键机理为H2O*中的O与邻甲酚—OH上的H存在氢键作用,影响表面物种的吸附稳定性,从而可能影响反应性能,但H2O*并未直接参与邻甲酚加氢脱氧反应;而氢传递机理则为H2O*转移1个H到邻甲酚脱下的OH*上,形成新的H2O*物种和—OH基团,H2O*直接参与反应,从而可能影响反应性能。图10为H2O*以不同机理参与邻甲酚HDO反应的初态、过渡态、终态最优结构。其反应能及活化能垒计算结果见表7。根据DFT计算结果可以看出,在Fe(211)表面上,当H2O*通过H-shuttling机理传递1个H原子而参与反应,相对于表面没有H2O*物种的情况,邻甲酚加氢脱氧生成甲苯反应能垒有所增加;当H2O*通过H-bonding机理仅稳定在邻甲酚附近而不参与加氢脱氧反应时,对CAr—O键断裂能垒几乎没有影响,进一步加氢,活化能垒略有升高。而在Pd-Fe(211)表面上,当H2O*通过H-shuttling机理参与反应,相对于表面没有H2O*物种的情况,邻甲酚加氢脱氧生成甲苯两步基元反应能垒均降低;当H2O*通过H-bonding机理仅稳定在邻甲酚附近而不参与加氢脱氧反应时,CAr—O键断裂反应能垒升高了0.12 eV,后续加氢活化能垒基本没有影响。当Pd-Fe(211)表面上存在3个吸附态H*时,H2O*通过H-shuttling机理参与反应(见图10(e)),与表面无H2O*的情况相比,邻甲酚直接脱羟基生成C7H7*中间体的活化能垒变化不大(见表7)。对于C7H7*中间体物种的进一步加氢转化生成甲苯,随着H*表面覆盖度的增加,加氢活化能垒随之降低,由0.54 eV降至0.47 eV,(见表7)。由此可见,当表面H2O*物种存在,且其以氢传递形式参与反应时,能够进一步促进Pd-Fe(211)表面上邻甲酚加氢脱氧生成目标产物甲苯。
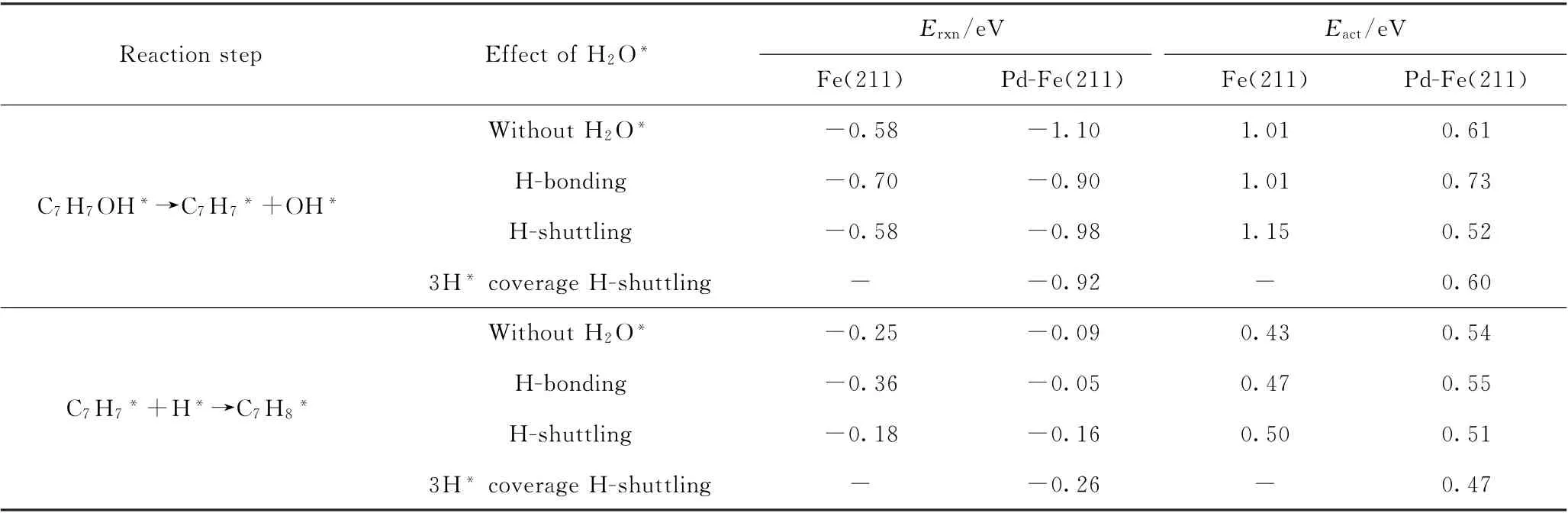
表7 H2 O*存在下Fe(211)及Pd-Fe(211)表面上邻甲酚直接脱羟基再加氢生成甲苯的反应能(E rxn)和活化能垒(E act)Table 7 Reaction energies(E rxn)and activation barriers(E act)associated with direct dehydroxylation of o-cresol followed by hydrogenation to produce toluene on Fe(211)and Pd-Fe(211)surfaces in the presence of surface H2 O*species

图10 H2 O*及3H*存在下Fe(211)及Pd-Fe(211)表面上邻甲酚直接脱羟基再加氢生成甲苯的反应初态(IS)、过渡态(TS)、终态(FS)的最优结构Fig.10 Optimized configurations of initial(IS),transition(TS)and final states(FS)associated with the direct dehydroxylation of o-cresol followed by hydrogenation to produce toluene on Fe(211)and Pd-Fe(211)surfaces in the presence of H2 O*and 3H*species
在加氢脱氧反应条件下,Fe催化剂表面可能会由于生成含氧物种而被氧化,而掺杂贵金属Pd能够更容易解离H2,增加表面H*物种的覆盖度,作为提高反应速率的途径来帮助清理Fe催化剂表面并调节Fe催化剂的相态,抑制其被氧化。计算了Fe(211)和Pd-Fe(211)表面上OH*+H*→H2O*的反应能和活化能垒。在Fe(211)上,Eact=1.32 e V,Erxn=0.97 e V;在Pd-Fe(211)上,Eact=1.11 e V,Erxn=0.75 e V,该结果表明掺杂Pd的Fe催化剂上更容易生成H2O*。在Pd-Fe(211)上,Pd的加入能够提供额外的H2吸附位点,促进其活化解离形成表面H*,在邻甲酚转化过程的加氢反应中具有高活性。此外,Pd与Fe的相互作用使表面H2O*物种更容易形成,其以氢转递机理参与邻甲酚的CAr—O键断裂反应,进一步促进目标产物甲苯的生成。
本研究的理论指导作用在于,从反应机理层面理解Fe基双金属催化邻甲酚加氢脱氧的催化剂构-效关系,以及Pd掺杂和H2O*参与反应对Fe催化剂活性和产物选择性的潜在影响。阐明双金属协同催化的微观本质,Pd有利于促进H2分子活化解离,提供了更多的表面H*物种,从而提高反应活性;Fe有利于促进脱氧反应高选择性生成芳烃产物。
5 结 论
通过DFT计算,系统地研究了Fe(211)表面上邻甲酚发生加氢脱氧反应生成苯酚及甲苯2种产物的反应机理,并揭示了Pd掺杂以及H2O*对反应性能的影响。得到如下结论:
(1)Fe(211)及Pd-Fe(211)表面上邻甲酚均以苯环与表面相互作用发生吸附,优于通过—OH基团进行吸附。在Fe(211)表面上,邻甲酚脱—OH生成甲苯选择性高于脱—CH3生成苯酚。在Fe(211)中掺杂Pd能够降低邻甲酚在催化剂表面上的吸附能。
(2)Pd的加入使邻甲酚在Fe表面上发生CAr—O键断裂再加氢生成甲苯的活化能垒降低。尤其当掺杂的Pd原子距离邻甲酚的羟基基团较远时,其脱羟基活化能垒显著降低,更有利于甲苯产物的生成。在Pd掺杂的存在下,H2分子易于活化和解离,表面H*物种覆盖度增加,影响关键表面物种的吸附能及其进一步转化,导致邻甲酚加氢脱氧反应速率加快。Pd掺杂Fe催化剂表现出对生成目标产物甲苯较好的活性和选择性。
(3)加氢脱氧过程中形成的表面H2O*物种易于以氢传递机理参与反应,使邻甲酚在Fe-Pd催化剂表面上选择性生成甲苯的活化能垒进一步降低,从而提高其选择性。
