藤黄酸类成分的硅胶干柱层析法分离及其酰胺新实体的合成*
2021-08-20周采菊高红刚郭永恩李爱华王保国
周采菊 高红刚 郭永恩△ 李爱华 刘 帅 王保国△
(1济宁医学院药学院;2日照市质量检验检测研究院,日照 276826;3济宁医学院临床医学院,济宁 272013)
中药材藤黄主要活性成分藤黄酸(GA)具有抗肿瘤、抗病毒、抗炎、抗氧化、抗菌及杀虫等生物活性[1];新藤黄酸(GNA)对多种肿瘤细胞株有抑制作用[2-3],且可加强化疗药物对某些肿瘤细胞敏感性[4]。它们均能选择性地攻击癌细胞而对正常造血系统和白细胞影响较小,但水溶性差 (1L水仅能溶解13mg GA)。目前GA、GNA的较高毒性、低水溶性、较差选择性、短半衰期等非成药性,限制了它们在抗癌领域的应用[5-6]。本实验通过从藤黄中提取分离GA和有创新性地从残余物中干柱逆流梯度层析法分离GNA,制备了N-(3-氯-4-氟苯基)藤黄酰胺、N-(3-氯-4-氟苯基)新藤黄酰胺和N-[2-(N’,N’-二甲氨基)乙基]藤黄酰胺3种新分子实体以求能改善它们的成药性。
1 材料与方法
1.1 主要试剂
2-(N’,N’-二甲氨基)乙胺购自阿拉丁公司;N,N’-二异丙基碳二亚胺(DIC)购自北京偶合科技有限公司;3-氯-4-氟苯胺购自AccelaChemBio.Co.Ltd;对二甲氨基吡啶(DMAP)购自武汉远成赛创科技有限公司;石油醚(b.p.60℃~90℃)购自天津市富宇精细化工有限公司;薄层层析硅胶H、硅胶GF254均购自青岛海洋化工有限公司;藤黄购自安徽亳州药材市场;硅胶GF254薄层板由本课题组铺制。
1.2 方法
1.2.1GA提取分离[7]取10g藤黄粉末用丙酮(30mL)超声提取18min倾出上清液,残渣加入丙酮(25mL)重复以上操作。至第4次提取后取少量丙酮溶解痕量残渣,与GA、GNA对照品溶液同板共薄层检测(TLC,展开剂石油醚-乙醇-三乙胺-乙酸乙酯v/v 1.2∶0.5∶0.2∶0.3)无GA、GNA斑点。其它同胡钟操作[7]得纯GA固体,ESIMS m/z 651[M+Na+],可直接用于本文下述反应。
1.2.2GNA的分离 1)制备含GA、GNA粗品的硅胶拌样。滤除GA吡啶盐沉淀,主要含GA、GNA粗品的母液,制备同胡钟操作[7]。减压蒸除吡啶后的2.0g残余物0.5%盐酸调pH 3~4后乙酸乙酯提取(9mL×4),至水层与GA、GNA对照品同板共薄层检测(展开剂石油醚-乙醇-三乙胺-乙酸乙酯v/v 1.2∶0.5∶0.2∶0.3)无GA、GNA斑点。合并提取液水洗(3mL×3) 分出有机层无水硫酸镁干燥1h,滤液减压浓缩后所得残余物溶于尽可能少的丙酮并加尽可能少的200目硅胶使没过液面,旋转蒸发器蒸除溶剂得粗品硅胶拌样(含60%左右的GA、GNA粗品)用于GNA的分离。
2)逆流干柱层析法从以上硅胶拌样中分离高纯度的GNA[8]。①装柱。如专利文献[8]所示,底端不带活塞、上端非磨口的带透砂板的层析柱中依次填充适量以上拌样粗硅胶、硅胶H[硅胶高度∶内径>8∶1,硅胶H与待分离样品重量比 (80~120)∶1]后,关闭洗脱剂装置部分的底部活塞并水泵抽实。②柱层析。关闭抽气部分、适度打开装洗脱剂分液漏斗的底部活塞,依次以适量体积的石油醚-乙醇-三乙胺-乙酸乙酯 v/v 6∶2∶1∶1.5体系和石油醚-乙醇-三乙胺-乙酸乙酯 v/v 4∶2∶1∶2体系梯度洗脱,按洗脱剂前沿1~3cm/min高度的上升速度开始干柱逆流柱层析直至洗脱剂前沿跑过硅胶H最上端、原拌样中样品在硅胶H柱上分出数条色带。③样品处理。取下色谱柱并底端连接打气筒小心把硅胶吹出到干净托盘中,把与薄层层析中GNA对应的谱带均匀分成3~5段,且每段取样TLC(展开剂石油醚-乙醇-三乙胺-乙酸乙酯v/v 1.2∶0.6∶0.2∶0.1)法检测其中GNA的纯度。分别合并以上含GNA纯品、较纯品的硅胶H,95%乙醇洗脱各段硅胶中的GNA至完全,洗脱液合并浓缩、称重后HPLC法检测纯度93%,ESI-MSm/z 631[M+H+]。
1.2.3GA和GNA酸酰胺新实体的制备[9-10]1)N-(3-氯-4-氟苯基)藤黄酰胺的合成。25mL圆底烧瓶中依次取0.6mL 5.0mmol/L的GA二氯甲烷溶液(含3.0μmol GA)、0.72mL 5.0mmol/L的DIC二氯甲烷溶液(含3.6μmol DIC) 和0.6mL 1.0mmol/L的DMAP二氯甲烷溶液(含0.6μmol DMAP) 室温搅拌15min后,加入0.66mL 5.0mmol/L 3-氯-4-氟苯胺二氯甲烷溶液(含3.3μmol 3-氯-4-氟苯胺)。室温继续搅拌10h,至TLC(展开剂石油醚-乙醇-三乙胺v/v 4.5∶1∶1)检测反应液中GA斑点消失。加水淬灭反应后分出有机层,依次水洗(3mL)、1%盐酸 (2mL)、水洗(3mL)后分出有机相无水硫酸镁干燥。滤除干燥剂滤液减压浓缩后,粗产物经干法上样、石油醚-乙醇-三乙胺-乙酸乙酯v/v 8∶1.2∶1∶1作洗脱剂的硅胶H干柱层析即得N-(3-氯-4-氟苯基)藤黄酰胺样品。ESI MS m/z 756[M+];1H NMR(CDCl3,400MHz)δ:12.70(s,1H,6-OH),8.01(s,1H,NH),7.56(d,J=7.1Hz,1H,10-H),7.30~7.46(m,3H,Ph-H),6.77~6.83 (m,1H,27-H),6.62(d,J=12Hz,1H,4-H),5.01~5.08(m,1H,37-H),3.46~3.51(m,1H,11-H),3.27~3.36(m,1H,31a-H),3.16~3.24(m,1H,31b-H),2.93(d,J=32Hz,2H,26-H),2.66~2.75(m,1H,32-H),2.54(d,J=9.4Hz,1H,22-H),2.28~2.35 (dd,J=13.4,4.7Hz,1H,21a-H),1.98~2.07(m,2H,36-H),1.31~1.81(m,28H)。见图1。
2)N-(2-N’,N’-二甲氨基)乙基藤黄酰胺的合成。25mL圆底烧瓶中依次取0.6mL 5.0mmol/L的GA二氯甲烷溶液(含3.0μmol GNA)、0.72mL 5.0mmol/L的DIC二氯甲烷溶液(含3.6μmol GNA) 和0.6mL 1.0mmol/L的DMAP二氯甲烷溶液(含0.6μmol DMAP) 室温搅拌15min后,加入0.50mL 10.0mmol/L(5.0μmol)N’,N’-二甲基乙二胺(DMEA)的二氯甲烷溶液。室温继续搅拌8h,至TLC(展开剂石油醚-乙酸乙酯v/v 4∶1)检测反应液中GA斑点消失。加水淬灭反应后分出有机层,水洗(2.5mL×2)后分出有机相无水硫酸镁干燥。滤除干燥剂滤液减压浓缩后,粗产物经干法上样、依次石油醚-乙酸乙酯 v/v 10∶1和6∶1作洗脱剂的硅胶H干柱层析即得N-[2-(N’,N’-二甲氨基)乙基]藤黄酰胺样品ESI MS m/z698 (M+);1H NMR(CDCl3,400MHz) δ:12.70(s,1H,6-OH),8.01(s,1H,NH),7.56(d,J=7.1Hz,1H,10-H),6.77~6.83(m,1H,27-H),6.62(d,J=12 Hz,1H,4-H),5.01~5.08(m,1H,37-H),3.46~3.51(m,1H,11-H),3.27~3.36(m,1H,31a-H),3.16~3.24(m,1H,31b-H),2.93(d,J=32Hz,2H,26-H),2.66~2.75(m,1H,32-H),2.54(d,J=9.4Hz,1H,22-H),2.28~2.35(dd,J=13.4,4.7Hz,1H,21a-H),1.98~2.07(m,2H,36-H),2.40(dd,2H,CH2),2.26(dd,2H,CH2),2.20~2.23(s,6H,2CH3),1.31~1.81(m,28H)。见图1。
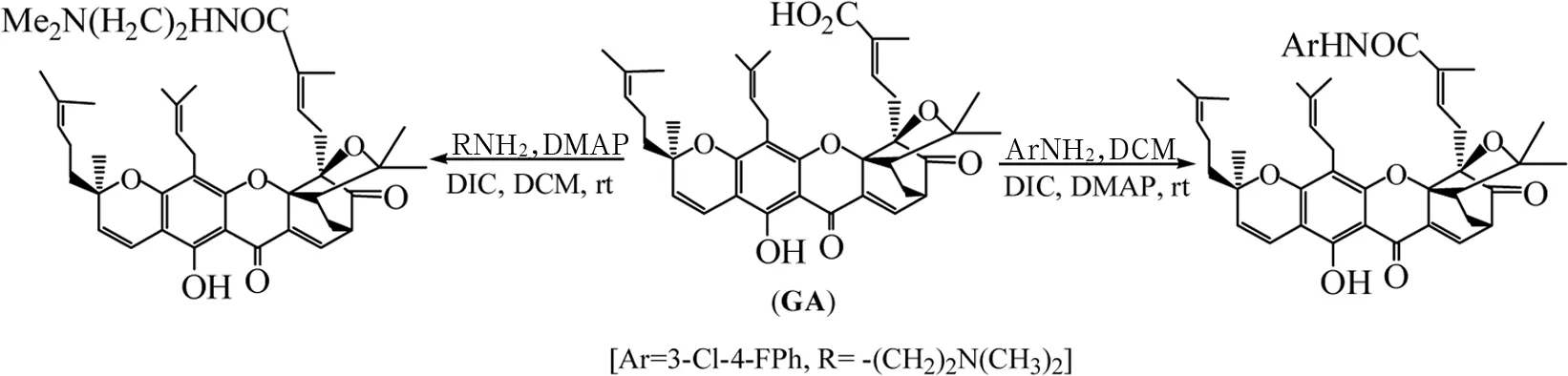
图1 N-芳基/烷基藤黄酰胺制备反应式
3)N-(3-氯-4-氟苯基)新藤黄酰胺的合成。25mL圆底烧瓶中依次取0.6mL 5.0mmol/L的GNA二氯甲烷溶液(含3.0μmol GNA)、0.66mL 5.0mmol/L的DIC二氯甲烷溶液(含3.3μmol DIC)和0.6mL 1.0mmol/L的DMAP二氯甲烷溶液(含0.6μmol DMAP) 室温搅拌15min后,加入0.72mL 5.0mmol/L 3-氯-4-氟苯胺二氯甲烷溶液(含3.6μmol 3-氯-4-氟苯胺)。室温继续搅拌11h至TLC(展开剂石油醚-乙醇-三乙胺-乙酸乙酯v/v 4∶1)检测反应液中GNA斑点消失。加水淬灭反应后分出有机层,依次水洗(3mL)、1% 盐酸(2mL)、水洗(3mL)后分出有机相无水硫酸镁干燥。滤除干燥剂滤液减压浓缩后粗产物经干法上样、依次石油醚-乙酸乙酯 v/v 10∶1和6∶1作洗脱剂的硅胶H干柱层析,即得N-(3-氯-4-氟苯基)新藤黄酰胺样品。ESI MS m/z758 (M+);1H NMR(CDCl3) δ:12.71(s,1H,6-OH),8.01(s,1H,NH),7.70~7.58(m,3H,Ph-H),7.53(d,J=7.1Hz,1H,10-H),6.43~6.46(m,1H,27-H),5.84(m,1H),5.20(t,1H,J=7.1,7.0Hz,3-H),5.02~5.08(m,1H,37-H),3.49(dd,1H,J=4.6,6.7Hz,11-H),3.17~3.36(m,5H),2.88(dd,1H,J=5.6,15.7Hz),2.48(d,J=9.1Hz,1H),2.30(dd,J=4.6,13.4Hz,1H),1.97~2.15(m,4H),1.31~1.81(m,26H)。见图2。

图2 N-芳基新藤黄酰胺制备反应式
2 结果和讨论
2.1 硅胶H逆流干柱梯度层析技术的优势及洗脱剂的确定
本文使用的硅胶H逆流干柱梯度层析技术分离层带窄而整齐、分离度大、可获得较高柱效,与传统湿法硅胶柱色谱相比有分离操作简便、耗时短、溶剂消耗量小、分离效率高等优点。柱层析前用硅胶GF254薄层层析法找出GNA斑点与其它斑点Rf差值较大、成点性好的展开剂,按极性增大顺序直接用作硅胶H柱逆流梯度层析中的洗脱剂。
2.2 藤黄粉提取溶剂的确定
从藤黄中提取GA、GNA时用丙酮代替甲醇作提取溶剂,能降低操作时的毒性且以上2种有效成分在丙酮中稳定性更大。
2.3 藤黄粉和新藤黄酸酰胺的设计理念
4-氟-3-氯苯胺和2-(N’,N’-二乙氨基)乙胺,分别是第二代、不可逆的EGFR酪氨酸激酶抑制剂达克替尼、可口服的多靶点酪氨酸激酶抑制剂苏尼替尼中的结构片段。GA、GNA结构中引入4-氟-3-氯苯胺和结构相似的2-(N’,N’-二甲氨基)乙胺,可能降低毒性并提高抗肿瘤活性,且引入2-(N’,N’-二乙氨基)乙胺片段还可能改善它们的水溶性,均可改善其成药性。
2.4 藤黄酰胺类制备中缩合试剂的选择
酰胺类的药物合成方法按上述操作N,N’-二环己基碳二亚胺(DCC)与1-羟基苯并三唑(HOBt)联合催化GA、GNA与脂肪胺的酰胺化,但与芳胺酰胺化时只得到中间产物活性酯。后改用DIC与DMAP按本文操作联合催化酰胺化,才成功得到N-芳基取代的藤黄酰胺和新藤黄酰胺。
以上GA、藤黄酰胺制备均以mmol/L浓度、μmol/L级的量进行。如其它条件不变而使用高浓度的相应试剂进行上述操作,反应速度则会增大。
利益冲突:所有作者均申明不存在利益冲突。
