伴癫痫性痉挛及局灶性发作等多种发作形式的Menkes病1例报告
2021-08-05王春利乔雪竹侯晓华姚丽芬
赵 蕊, 王春利, 乔雪竹, 刘 菲, 侯晓华, 姚丽芬
Menkes病是一种罕见的先天性铜代谢异常疾病,为X连锁隐性遗传性疾病,由编码铜转运蛋白(ATP7A)的基因出现致病性突变引起。致病性ATP7A基因突变的分子后果使铜转运受损,进而导致关键的含铜酶(多巴胺b羟化酶和细胞色素c氧化酶)缺乏,因此导致涉及这些酶的代谢途径障碍。临床特点主要包括:进行性神经系统变性、结缔组织异常及特征性的毛发改变,血清铜及铜蓝蛋白水平降低,头部血管成像(MRA)显示动脉扭曲、螺丝锥样改变,常伴有癫痫发作。现报道1例典型Menkes病患儿的电-临床-影像学特征,并结合文献资料对其进行分析。
1 临床资料
1.1 患儿,男,8个半月。因“瞬间环抱样动作、有时伴哭闹1 w”于2020年9月11日来我院就诊。1 w前患儿无明显诱因出现四肢瞬间环抱样动作,有时动作结束后,伴哭闹。于困倦或睡醒后发作次数增多。既往史:患儿4月龄时因发育迟缓,行基因检查。基因为:ATP7A基因异常,染色体位置:chrX:77301954 c4390A>G(半合子变异),父亲未发现变异,母亲杂合(见图1),临床考虑为Menkes病,未给予治疗。家族史:正常。查体:神清,生命体征平稳,体重8.9 kg,头围44 cm.特征性面容:皮肤白,毛发稀疏、色浅,前头部发直、后头部发卷曲,枕骨隆起;双下颌,面颊饱满,嘴宽;全身皮肤松弛,腹股沟疝,重度躯干肌张力减低,头部控制差,抬头无力。心肺腹查体未见异常,外生殖器正常。颅神经检查未见异常。四肢肌力检查不配合,未见自主活动,四肢肌张力明显低下,双侧膝腱反射对称,腱反射活跃,双侧巴氏征(+)。辅助检查:MRI:双基底节异常信号,髓鞘化略落后,双额颞顶脑沟裂略宽深,脑室系统略大。MRA:双侧颈内动脉颅内段血管迂曲,左侧大脑中动脉显影淡,管腔明显狭窄,远端分支较对侧稀少(见图2)。脑电图结果:背景活动:枕区未见符合年龄发育节律,弥漫中高波幅1.5~3.5 z混合频率慢活动。发作间期放电:清醒期显示多灶棘波、尖波、棘慢、尖慢波,有失律倾向,棘波以双侧颞区明显;左额-中央期间断出现波幅5~8 c/s电活动,其间杂以棘慢活动。睡眠期呈高度失律图形,表现为不规则杂乱的高波幅慢波、棘波、多棘波及慢活动,较持续或间断性出现,伴多灶棘波,仍以双颞区明显,双颞枕区可见棘波其上附着高频快节律波。发作期:癫痫性痉挛、强直痉挛发作混合发作(孤立或成串);癫痫性痉挛+局灶性发作混合类型(见图3、图4)。生化检查:血清铜离子:2.05 μmol/L,铜蓝蛋白:0.05 g/L。

图1 患儿基因检查结果:ATP7A基因 c.4390A>G,错义突变
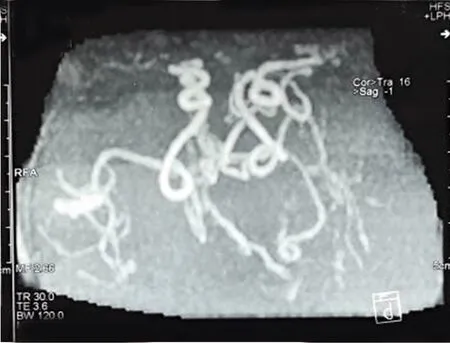
图2 MRA:双侧颈内动脉颅内段血管迂曲,左侧大脑中动脉显影淡,管腔明显狭窄,远端分支较对侧稀少

图3 强直痉挛发作:一次痉挛后,肌电出现强直成分

图4 混合发作:一次痉挛发作后左额、中央区快节律起始的局灶性发作,同期患儿表现为动作停止、头眼向左侧略偏斜
1.2 诊断 患儿男性,特殊面容,肤白、毛发稀疏卷曲,全身肌张力减低,腹股沟疝。发育迟缓,以癫痫性痉挛及局灶性癫痫发作就诊。综合临床表现、辅助检查、基因诊断,临床诊断为Menkes病。
1.3 治疗过程及随访 临床给予:喜保宁500 mg,bid po;开浦兰:200 mg,bid po;组氨酸铜:1 ml皮下注射。于2个月后进行随访,患儿头围、体重未增长,仍有严重肌张力减低及神经系统发育迟滞,双眼可追随物体,家属自述患儿近一个月内癫痫发作明显减轻。该患儿于2020年10月9日再次进行全基因组水平拷贝数变异(CNV)检测,结果:未检测到可以明确解释患者表型的致病性或疑似致病性拷贝数变异(CNV)。
2 讨 论
Menkes病是一种X连锁的隐性疾病,是一种致命的铜代谢异常导致的多系统疾病,其特征是结缔组织异常、进行性神经退行性病变。癫痫发作是该病的主要临床特征之一,癫痫的病理基础考虑与铜介导的NMDA受体功能损伤,导致神经递质功能障碍、能量代谢及兴奋性毒素等共同作用有关。大多数患者在2~3个月大时会出现癫痫发作。癫痫过程常分为3个阶段:早期为局灶性阵挛性发作及癫痫持续状态,中期常为癫痫性痉挛,晚期为多灶性肌阵挛和强直发作[1]。在Menkes病国内报道中,可见数例癫痫性痉挛发作及早期起病的局灶性阵挛发作及癫痫持续状态的报道。本例患儿出生正常,出生后发育正常。4个月大时出现运动功能异常,8个月大时出现癫痫性痉挛发作[2],脑电图检查显示背景脑电活动异常,未见睡眠结构出现。清醒期及睡眠期均有间断高度失律(睡眠期明显)、多灶棘波。本例患儿监测中出现多种癫痫发作形式:癫痫性痉挛、强直痉挛发作以及典型痉挛与局灶性发作的混合发作形式。其中伴有典型癫痫性痉挛+局灶性发作的癫痫混合发作形式在Menkes病国内病例报道中为首次。根据患儿此次的癫痫发作特点,考虑目前患儿处于癫痫发作进程的中晚期阶段[3]。
根据突变等位基因的不同,Menkes病表型轻重有所不同,典型病例累及结缔组织、神经系统功能损害较重。患儿一般具有特殊面容,表现为肤白、毛发稀少而卷曲、枕骨凸出,高弓形上颚等,典型病例还有脐疝、腹股沟疝、关节活动过度、皮肤松弛、肌张力低下和进食问题、以及神经、和精神系统发育延迟、癫痫发作,本例患儿具有上述大部分特征。
迄今为止大约描述了170多种编码ATP7A蛋白基因的不同突变,约25%是大的缺失,从单个外显子到整个基因的缺失(前两个外显子除外)。并已描述约120种不同的ATP7A基因内突变[4],无义突变(16%),错义突变(33%),剪接位点突变(16%)和缺失/插入/重复(33%)。跨膜结构域中的错义突变,可能导致部分功能性蛋白质变体,表型相对较重。本例患儿ATP7A基因c4390A>G错义突变,目前尚无文献报道突变位点具有致病性,疑似良性变异,但本例患儿临床表型较重、基因型变异与表型特点不符,具体原因尚不清楚,不除外有其他基因位点出现变异尚未检出的可能,有待进一步研究验证。
有研究探讨了Menkes病的电临床特点以及核磁共振表现[5~8],癫痫发作类型包括多灶性阵挛性发作、肌阵挛性抽搐和强直性痉挛,所有患者的脑电图均明显异常。患儿前6个月以局灶性放电为主导,但其后特征性地出现了高度失律。MR成像显示所有患者均异常,最常见结果是脑萎缩和髓鞘延迟。上述报道的脑电图以及核磁共振表现与本病例相似,遗憾的是本例患儿无起病时(4个月)的脑电图,无法观察该患儿脑电图演变的过程。本病例中患儿颅内颈内动脉段迂曲以及左侧大脑中动脉狭窄,这与以往报道相似。目前认为Menkes病颅内血管迂曲的发病机制是由于铜依赖赖氨酰氧化酶缺乏导致动脉的内弹性膜、中膜和内膜层的弹性蛋白纤维的缺陷,从而引起动脉的曲折、延长、扩张或狭窄。
以往报道的Menkes病患者,妊娠期通常并无特殊异常,新生儿期常以腹泻、低血糖、长期黄疸和发作性体温过低为特征,这些症状通常延续到整个生命期,本例患儿孕产史均正常,发病前未发现特殊异常。
该病是罕见的X连锁隐形遗传病。熟练掌握该病的病因、家族史、临床表现、影像学特征,有助于对该类疾病早期识别和诊断。目前该病无特殊有效治疗办法。对症支持治疗是主要办法。胃肠外组氨酸铜替代治疗已被证实是安全有效的,但不能缓解已有的神经系统症状,需终身治疗[9]。
Menkes病具有特征性面容、不同时期有特征性的脑电图模式及显著的影像学特征。基因检测是诊断该病的金标准,但是在基因检测结果出来之前,掌握特殊面容、脑电图及影像学特点,对该病的早期诊断有非常重要的意义,可为早期精准治疗提供可靠的依据。
