花生荚果与种子相关性状QTL定位及与环境互作分析
2021-08-05孟鑫浩张靖男崔顺立CharlesChen穆国俊侯名语杨鑫雷刘立峰
孟鑫浩 张靖男 崔顺立 Charles Y. Chen 穆国俊 侯名语 杨鑫雷,* 刘立峰,*
花生荚果与种子相关性状QTL定位及与环境互作分析
孟鑫浩1张靖男1崔顺立1Charles Y. Chen2穆国俊1侯名语1杨鑫雷1,*刘立峰1,*
1华北作物改良与调控国家重点实验室/ 河北省种质资源重点实验室/ 河北农业大学, 河北保定 071001;2奥本大学作物、土壤与环境科学系, 美国奥本 36849
花生荚果、种子性状与产量紧密相关, 是重要的农艺性状。为挖掘与荚果、种子性状紧密连锁的分子标记, 本研究以大果品种冀花5号和小果美国资源M130组配衍生的315个家系RIL8群体为材料, 利用SSR、AhTE、SRAP和TRAP等标记构建了一张包含363个多态性位点的遗传连锁图谱。该图谱共包含21个连锁群, 总长为1360.38 cM, 标记间平均距离为3.75 cM。利用完备区间作图法对2017—2018年5个环境的荚果、种子相关性状进行数量性状基因座(quantitative trait locus, QTL)分析, 共鉴定到97个与荚果、种子性状相关的QTL, 可解释的表型变异为2.36%~12.15%, 分布在A02、A05、A08、A09、B02、B03、B04、B08和B09等9条染色体上。其中, 9个与荚果长相关, 13个与荚果宽相关, 14个与荚果厚相关, 11个与种子长相关, 14个与种子宽相关, 13个与种子厚相关, 13个与百果重相关, 10个与百仁重相关; 4个主效QTL分别为和, 可解释的表型变异分别为10.02%、11.06%、12.15%和11.97%; 45个稳定表达的QTL在3个以上环境可被重复检测; 连锁群A02、A08、B02、B04和B08上存在QTL聚集区。另外, 检测到15对上位性QTL, 可解释的表型变异为10.23%~51.84%。研究结果将为花生荚果、种子性状的分子标记辅助育种提供重要的理论依据。
花生; 荚果; 种子; QTL; QTL×E
花生(L.)又名落花生, 起源于南美洲, 是热带和亚热带地区种植广泛的油料作物和经济作物, 也是人类食用油的主要来源之一[1], 产量相关性状遗传研究广受关注。花生荚果、种子性状属于重要的产量性状, 是典型的数量性状, 受多基因与环境共同调控, 其遗传规律十分复杂[2]。研究数量性状的遗传机制为花生分子育种提供了新的可能, 而单靠常规育种已无法满足生产上的需要, 需借助分子标记辅助选择技术[3]。分子标记可以对与产量性状密切相关的数量性状进行选择, 提高选择的准确性, 从而加快花生育种进程, 对培育高产花生新品种具有重要意义。数量性状定位是研究数量性状的主要方法之一, 亦是揭示花生荚果、种子相关性状遗传规律的重要途径[4]。RIL群体属于永久性定位群体, 具有遗传稳定、QTL定位及遗传效应分析准确等优点[5]。
利用RIL群体进行多环境花生荚果、种子性状QTL定位的研究已有进展。Luo等[6]利用“徐花13×中花6号”重组自交系群体, 在4个环境下检测到33个与荚果性状相关的QTL, 主要分布在A05、A07、A08等染色体上。Wang等[7]利用“ZH16×sd-H1”构建的RIL群体作为研究材料, 在3个环境下获得30个荚果相关的QTL, 主要分布在B06和B07染色体上。Luo等[8]利用构建的栽培种花生遗传图谱, 在4个环境下重复检测到42个与荚果性状相关的QTL, 主要分布在A05、A06、A09和B05染色体上。李英杰[9]利用已构建的遗传图谱, 对10个产量相关性状进行QTL定位分析, 共检测到8个主效QTL, 单个QTL的PVE为5.66%~28.05%。李振动等[10]以“远杂9102×徐州68-4”衍生的RIL群体为材料, 2年间共检测到41个数量性状位点, 其可解释的表型变异(phenotypic variation explained, PVE)为3.14%~ 18.27%, 有6个位点在2个环境下被稳定检测到。吕维娜等[11]利用“白沙1016×”构建的RIL群体为试验材料, 在3个不同的环境下定位到72个与百果重、百仁重等农艺性状相关的数量性状位点。成良强[12]以“富川大花生×ICG6375”衍生的F2群体为研究材料进行QTL定位, 获得68个与荚果长、荚果宽、种子长、种子宽、百果重和百仁重等产量性状相关的QTL。Shirasawa等[13]以2个F2群体为材料, 检测到23个QTL与荚果、种子性状相关, 其PVE为4.8%~28.2%。
目前, 花生荚果、种子相关性状QTL在不同研究间仍存在较大差异, 多环境下稳定的QTL仍较少。因此, 本研究以大果型花生品种冀花5号和小果型美国种质资源M130组配衍生的包含315个家系的RIL群体为材料, 利用SSR、SRAP、TRAP等分子标记构建花生遗传连锁图, 并利用完备区间作图法对5个环境下的8个荚果、种子相关性状进行QTL定位及与环境的互作效应分析。鉴定出的QTL将为荚果和种子性状的遗传解析、图位克隆和遗传改良提供理论参考。
1 材料与方法
1.1 试验材料
以大果型花生品种冀花5号为母本, 小果型美国花生种质资源M130为父本(图1), 采用单粒传法, 构建包含315个家系的F8重组自交系群体为研究材料。2017年分别种植在河北省邯郸市大名县(DM, 35º57′N & 115º09′E)和河北省保定市清苑区(QY, 38º40′N & 115º30′E)。2018年在河北省唐山市迁安市(QA, 39º99′N & 118º70′E)、河北省邯郸市大名县和河北省保定市清苑区等地分别种植亲本和群体。每个家系种植1行, 行长1.5 m, 行距为0.5 m, 株距为0.17 m。每行种植10株。随机区组设计, 2次重复, 常规田间管理。试验材料收获晒干后, 参考姜慧芳等[14]的《花生种质资源描述规范和数据标准》对荚果长(pod length, PL)、荚果宽(pod width, PW)、荚果厚(pod thickness, PT)、种子长(seed length, SL)、种子宽(seed width, SW)、种子厚(seed thickness, ST)、百果重(hundred-pods weight, HPW)、百仁重(hundred-seeds weight, HSW)等8个产量相关性状进行表型鉴定。
白色指示条为标尺, 大小为1 cm。The white bar indicates 1 cm.
1.2 DNA提取及引物筛选
在田间取亲本和RIL群体各株系的幼嫩倒三叶, 采用改良SDS-CTAB法[15]提取花生基因组总DNA。利用3964对SSR标记和转座子引物(https:// peanutbase.org/)、238对SRAP引物[16]、155对TRAP引物[17]等对双亲进行多态性筛选。PCR体系及PCR扩增的反应程序参考崔顺立等[18]的试验方法进行, PCR产物通过8%非变性聚丙烯酰胺凝胶电泳进行分子标记的多态性检测。
1.3 基因型统计及连锁图谱构建
群体基因型采用母本带型记为“a”, 父本带型记为“b”, 杂合带型记为“h”的标记方法进行统计。带型模糊不清或数据缺失使用“-”替代。群体基因型通过JoinMap 4.0[19]进行连锁分析, 设置步长为0.5, LOD > 3, 在LOD值3~10范围内将所得到的标记进行分组, 利用Kosambi函数将重组率转化为连锁距离[20]。采用Mapchart 2.3[21]绘制遗传连锁图谱。构建好的遗传连锁图谱与Peanut genome resource (http://peanutgr.fafu.edu. cn/Maps_traits.php)网站上公布的包含1954个标记位点的整合图谱进行共线性分析。
1.4 表型统计及QTL定位
采用GraphPad Prism 8 (https://www.graphpad. com/scientific-software/prism/)对2年5个环境的群体表型值进行统计分析, 估计基因型与环境互作的效应[22], 计算广义遗传力[23]。利用QTL IciMapping 4.2[24]中的ICIM-ADD方法对不同环境下各性状进行QTL定位和效应估计, 采用ICIM-EPI方法评估QTL与环境之间的效应。一般来说, 可解释的表型变异(phenotypic variation explained, PVE)大于10%的QTL被认为是主效QTL, 其他QTL被称为微效QTL[25]。QTL的命名以“”开头, 加上性状名称和染色体名称, 如果同一连锁群上出现2个或2个以上相同性状的QTL, 则在连锁群后面加上“.”和数字进行区分[11], 如在A08染色体上有2个与荚果长相关的QTL, 则分别命名为和。
2 结果与分析
2.1 表型统计分析
2.1.1 荚果和种子的表型及相关性分析 对2017—2018年花生亲本及其RIL群体各家系荚果长、荚果宽、荚果厚、种子长、种子宽、种子厚、百果重、百仁重等8个性状进行描述性统计分析(表1)和相关性分析(表2)。结果表明, 在5个环境中, 父母本在每个性状均表现出显著差异, RIL群体具有较大的变异范围, 且群体各性状的最小值和最大值均超过亲本, 说明各性状都具有正向超亲和负向超亲优势。群体表型值的偏度和峰度均趋于0, 且test均表现出不同程度的显著性, 说明各性状符合正态性(表1和图2), 适合QTL分析。相关性分析结果表明, 各个性状在同一环境下均表现为极显著的正相关关系(<0.001) (表2), 推测多个性状可能被定位到同一个QTL区间, 即“一因多效”。
2.1.2 方差分析 通过2017—2018年的8个荚果、种子相关性状方差分析(表3)可知, 同一环境下各性状在重复间不存在显著性差异(>0.05); 群体各家系的同一性状存在极显著差异(<0.01); 同一性状在不同环境下表现出极显著差异(<0.01); 群体基因型与环境互作(G×E)在不同性状间均呈现极显著差异(<0.01)。各性状遗传变异系数最小的为荚果长(GCV=12.87%), 最大的为百果重(GCV=17.54%); 荚果、种子相关性状均呈现中等遗传力(54.09%~67.50%), 说明各性状受环境影响较大。综合方差分析结果推测, 荚果、种子相关性状受微效多基因控制的可能性较大, 各性状可能会定位到多个QTL位点。
缩写同表1。Abbreviations are the same as those given in Table 1.
2.2 遗传连锁图构建与共线性分析
利用SSR、AhTE、SRAP和TRAP等分子标记对亲本进行多态性筛选, 获得640对条带清晰、多态性好的分子标记用于RIL群体基因分型。将分型后的基因型数据通过JoinMap 4.0软件(设置LOD=3.0~10.0)进行连锁分析, 获得1张包含21个连锁群的遗传连锁图谱(图3)。该图谱包含363个标记位点, 单条连锁群长度为39.59~101.05 cM, 包含4~50个分子标记, 标记间平均距离为3.75 cM。其中, 标记位点最少的染色体为B06 (4个), 标记位点最多的染色体为B09 (50个), 29个标记位点未匹配到染色体上, 命名为Unknown连锁群。与高密度图谱进行共线性分析(图4)可知, 84个标记与整合图谱符合, 分布在A01 (11个)、A02 (3个)、A04 (5个)、A05 (2个)、A07 (5个)、A08 (5个)、A09 (5个)、A10 (4个)、B01 (3个)、B02 (5个)、B03 (4个)、B04 (7个)、B05 (1个)、B06 (1个)、B07 (4个)、B08 (6个)、B09 (8个)、B10 (5个)染色体上。除A05染色体外, 其他染色体上的标记顺序均存在位置颠倒变化。此外, A03和A06染色体上的标记与整合图谱无共线性标记。
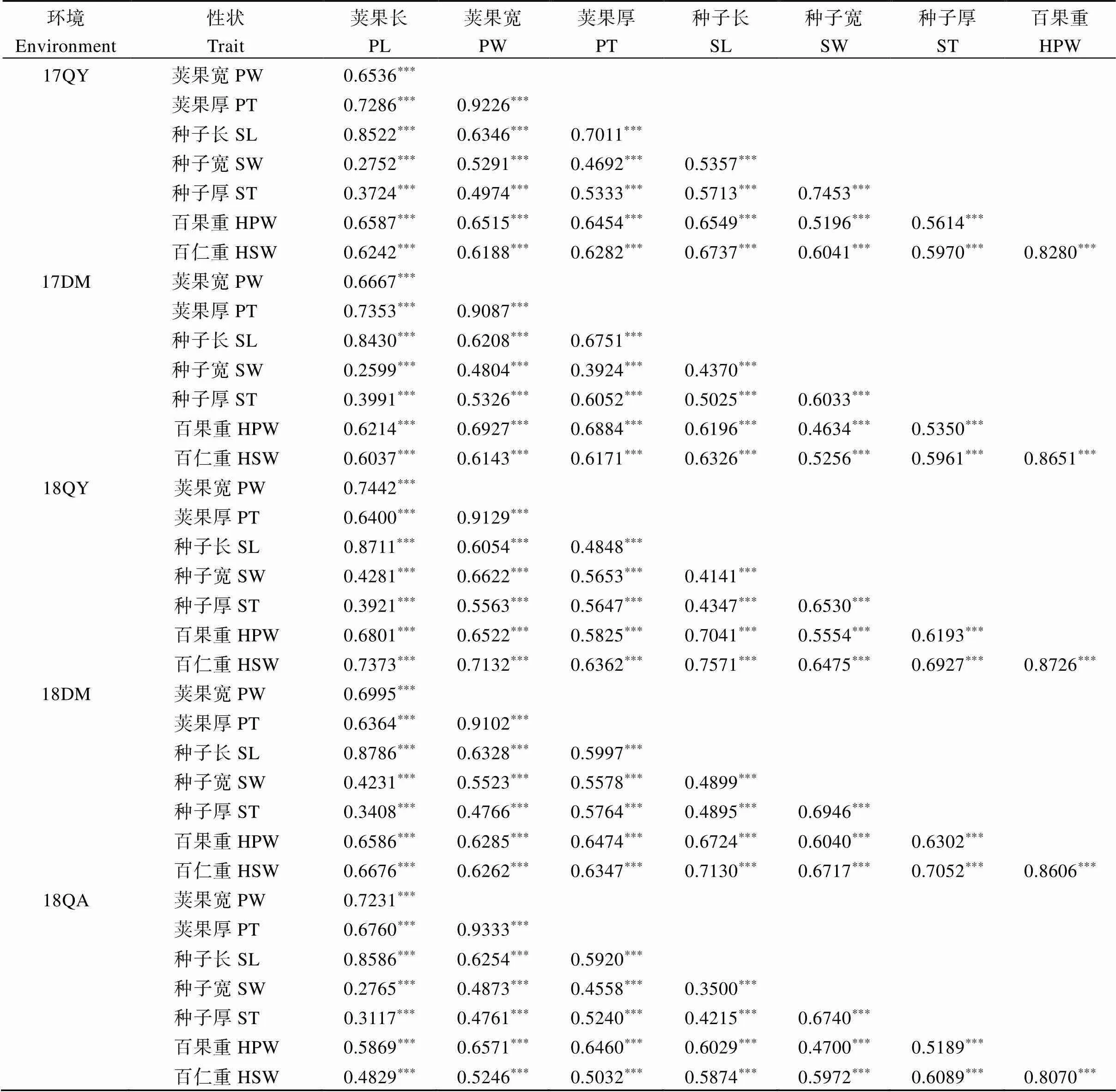
表2 不同环境下各性状的相关性分析结果
***表示在0.001水平显著相关。缩写同表1。
***: Significant correlation at< 0.001. Abbreviations are the same as those given in Table 1.

表3 RIL群体各性状方差分析及广义遗传力
ns代表差异不显著,*和**分别代表在0.05和0.01水平差异显著。缩写同表1。
ns: not significant;*and**represent significant difference at the 0.05 and 0.01 probability levels, respectively. Abbreviations are the same as those given in Table 1. GVC: genetic coefficient of variation.
缩写同表1。Abbreviations are the same as those given in Table 1.
2.3 QTL定位
2.3.1 加性QTL分析 荚果、种子性状QTL定位分析表明, 在2017—2018年5个环境下共检测到97个QTL (表4和图3), 分布在A02、A05、A08、A09、B02、B03、B04、B08等8条染色体上, 其贡献率在2.36%~12.15%, LOD值在2.52~11.01, 加性效应为-10.993~12.309。其中, 主效QTL有4个, 2个与荚果厚相关(和), 位于A08染色体HBAUAh177~AhTE0658标记区间, PVE分别为11.06% (LOD=11.01)和12.15% (LOD=9.44); 1个与荚果宽相关(), 位于A08染色体HBAUAh177~AhTE0658标记区间, PVE为10.02% (LOD=8.35); 1个与种子宽相关(), 位于B08染色体AHGS1286~TC20B05标记区间, PVE为11.97% (LOD=9.98)。58个QTL的加性效应值为正, 推测控制性状的基因可能来源于母本; 39个QTL的加性效应值为负, 推测控制性状的基因可能来源于父本。本研究共检测到9个QTL聚集区(图3), 分别为A02染色体的AHGS1163~AHGS1886标记区间、A08染色体上的Ah4-4~TC9B08、me3em14- 196~Ah4-4、AhTE0658~TC6H03、TC6H03~AhTE0477和HBAUAh177~AhTE0658标记区间, B02染色体的AHTE0398~CTW_NEW_38标记区间、B04染色体的T3em5-340~me1em3-75标记区间、B08染色体的AHGS1286~TC20B05标记区间, 涉及荚果、种子8个相关性状, 说明这些染色体区段上的基因可能存在“一因多效”的现象。另外, 本研究在3个以上环境重复检测到的稳定QTL有45个(表4和图3), 分布在A02、A08、B04和B08染色体上, 说明这些QTL受环境影响较小。
2.3.2 上位性QTL分析 对8个荚果、种子性状在5个环境下进行上位性QTL分析(表5), 共获得15对上位性QTL, 涉及荚果长、荚果宽、荚果厚、种子长、百果重和百仁重等6个性状, 其LOD值为5.09~8.05, PVE为10.23%~51.84%。其中, 控制荚果长、种子长和百仁重的上位性QTL各1对, 其LOD值分别为5.09、7.60和7.30, PVE分别为10.23%、11.61%、13.99%; 控制百果重的上位性QTL有2对, 其LOD值为8.05和5.04, PVE为15.36%、17.75%; 控制荚果宽的上位性QTL有2对, 其LOD值为5.33和5.54, PVE为11.95%~42.46%; 控制荚果厚的上位性QTL有8对, 其LOD值为5.01~7.66, PVE为26.30%~51.84%。
2.4 QTL与环境互作效应结果分析
2.4.1 加性QTL与环境互作分析 利用IciMapping 4.2软件加性QTL与环境的互作效应进行分析(表6), 共有6对与环境有互作效应的加性QTL, 与荚果长、种子厚和百果重相关的QTL各1个, 其加性效应值分别为-0.55、-0.37和0.36, 加性遗传贡献率分别为1.280%、0.624%和0.618%, 与环境互作的遗传贡献率分别为0.082%、0.059%和0.014%; 与种子长相关的QTL有3个, 其加性效应值为-0.642~-0.491, 加性遗传贡献率为1.10%~1.90%, 与环境互作的遗传贡献率为0.046%~0.159%。
2.4.2 上位性QTL与环境互作分析 由表7可知, 共有13对上位性QTL与环境有互作效应, 其中3对与荚果长相关, PVE为1.95%~2.65%, PVE (AAE)为0.007%~0.089%; 与荚果厚相关的QTL有6对, PVE为1.47%~2.81%, PVE (AAE)为0.007%~0.073%; 与种子长和百仁重分别相关的QTL各2对, 其PVE分别为1.286%、4.009%和2.417%、1.489%, PVE (AAE)分别为0.137%、0.024%和0.039%、0.019%。所有和环境互作的QTL, 在不同的环境下均表现出不同的互作效应。
3 讨论
花生荚果、种子性状与产量紧密相关, 是重要的农艺性状。本研究选用冀花5号和M130 2个花生品种作为亲本材料, 其生长发育和对环境的适应性均表现良好, 而且2个亲本间与荚果、种子相关的8个性状差异显著(<0.01)。在构建的RIL群体中, 荚果长、荚果宽、荚果厚、种子长、种子宽、种子厚、百果重和百仁重等8个性状的变异符合正态分布(图2), 且表现出较大的变异范围, 其最大、最小值均超过双亲, 这为构建花生遗传连锁图以及QTL分析奠定了坚实的基础。
一般认为, 主效QTL的贡献率大于10%, LOD值越大, QTL越稳定。本研究通过构建遗传连锁图谱,结合多年多环境的表型数据共检测到97个QTL, 其中, 在A08染色体AHTE0658标记附近检测到与百仁重相关的QTL, 与Lu等[27]的产量QTLs meta分析结果一致。然而, Lu等[27]在B08染色体AHGS2268~ AHGS494区间检测到2个与百果重相关的Meta QTL, 而本研究与百果重相关的QTL则是在B08染色体的AHGS1286~TC20B05区间, QTL标记区间的差异可能是由于图谱之间的标记种类不一致所造成。在与前人[13,28-32]的研究对比中发现, 前人重复检测到的QTL主要集中在A05染色体上, 而本研究定位到的QTL集中分布在A02、A08和B02染色体上, 并且具有较高的贡献率。此外, 4个主效QTL分别分布在A08染色体() HBAUAh177~AhTE0658区间和B08染色体上() AHGS1286~TC20B05区间上, 与Lu等[27]的研究结果不同, 说明这些QTL是全新的。本研究发现了9个QTL聚集区, 其中5个QTL聚集区分布在A08染色体上, 涉及所有的荚果、种子相关性状, 且在多个环境下能够被重复检测到, 表明所检测到的QTL是稳定的, 且在A08染色体上与荚果、种子性状相关的QTL鲜有报道。因此, A08染色体是研究花生荚果、种子相关性状十分重要的染色体之一, 可进一步进行分子标记的加密研究。
基因型×环境互作是作物数量性状的普遍属性和遗传育种改良的关注重点, 但在前人[27-32]的研究中未进行基因型×环境互作分析, 本研究共获得6个与环境互作的加性QTL和15对上位性QTL, 涉及荚果长、荚果厚、种子长、种子厚、百果重和百仁重等性状, 说明在花生荚果、种子相关性状中存在基因型×环境互作效应。因此, 在利用QTL改良作物品种时, 既需注重QTL的遗传主效应, 还需重视QTL与环境的互作效应[33], 上位性效应对数量性状亦有重要的作用[34]。随着分子标记的开发应用和QTL定位技术的不断发展, 在分子标记辅助育种中既应该考虑起主效作用的QTL, 又要考虑与其存在上位效应的QTL。然而, 同一性状的QTL 在不同定位群体和不同环境下可能表现不一致, 通过对QTL在不同环境背景下的研究, 有利于定位到受环境影响较小的QTL。不仅考虑到显性和上位性, 同时考虑到与环境的互作效应, 有助于提高分子标记选择的效率。
4 结论
本研究构建了1张包含363个标记位点、21条连锁群、覆盖长度为1360.38 cM的花生遗传连锁图谱。利用该图谱对2017—2018年5个环境下8个荚果、种子相关性状进行QTL定位, 获得97个QTL, 其中, 4个主效QTL分别为、、和。3个以上环境重复检测到的稳定QTL有45个, A02、A08、B02、B04和B08染色体上存在9个QTL聚集区。对8个荚果、种子相关性状在5个环境下进行上位性QTL定位分析, 检测到15对上位性QTL, 涉及荚果长、荚果宽、荚果厚、种子长、百果重和百仁重等6个性状。
[1] 禹山林. 中国花生品种及其系谱. 上海: 上海科技出版社, 2008. pp 353–465.
Yu S L. Chinese Peanut Varieties and Their Lines. Shanghai: Shanghai Scientific & Technical Publishers, 2018. pp 353–465 (in Chinese).
[2] Xiang D Q, Cao H H, Cao Y G, Yang J P, Huang L J, Wang S C, Dai J R. Construction of a genetic map and location of quantitative trait loci for yield component traits in maize by SSR markers., 2001, 28: 778–784.
[3] Varshney R K, Close T J, Singh N K, Hoisington D A, Cook D R. Orphan legume crops enter the genomics era!, 2009, 12: 202–210.
[4] 方宣钧. 作物DNA标记辅助育种. 北京: 科学出版社, 2001. pp 305–336.
Fang X J. Crop DNA Marker-assisted Breeding. Beijing: Science Press, 2001. pp 305–336 (in Chinese).
[5] Wang B H, Guo W Z, Zhu X F, Wu Y T, Huang N T, Zhang T Z. QTL mapping of yield and yield components for elite hybrid derived-RILs in upland cotton., 2007, 34: 35–45.
[6] Luo H Y, Guo J B, Ren X P, Chen W G, Huang L, Zhou X J, Chen Y N, Liu N, Xiong F, Lei Y, Liao B S, Jiang H F. Chromosomes A07 and A05 associated with stable and major QTLs for pod weight and size in cultivated peanut (L.)., 2018, 131: 267–282.
[7] Wang Z H, Huai D X, Zhang Z H, Cheng K, Kang Y P, Wan L Y, Yan L Y, Jiang H F, Lei Y, Liao B S. Development of a high-density genetic map based on specific length amplified fragment sequencing and its application in quantitative trait loci analysis for yield-related traits in cultivated peanut., 2018, 9: 827.
[8] Luo H Y, Ren X P, Li Z D, Xu Z H, Li X P, Huang L, Zhou X J, Chen Y N, Chen W G, Lei Y, Liao B S, Pandey M K, Varshney R K, Guo B Z, Jiang X G, Liu F, Jiang H F. Co-localization of major quantitative trait loci for pod size and weight to a 3.7 cM interval on chromosome A05 in cultivated peanut (L.)., 2017, 18: 58.
[9] 李英杰. 栽培种花生株高、分枝数及荚果性状QTL定位分析. 山东农业大学硕士学位论文, 山东泰安, 2016.
Li Y J. QTL Analysis for Plant Height, Total Branching Number and Pod Traits in Peanut (L.). MS Thesis of Shandong Agricultural University, Tai’an, Shandong, China, 2016 (in Chinese with English abstract).
[10] 李振动, 李新平, 黄莉, 任小平, 陈玉宁, 周小静, 廖伯寿, 姜慧芳. 栽培种花生荚果大小相关性状QTL定位. 作物学报, 2015, 41: 1313–1323.
Li Z D, Li X P, Huang L, Ren X P, Chen Y N, Zhou X J, Liao B S, Jiang H F. Mapping of QTLs for pod size related traits in cultivated peanut (L.)., 2015, 41: 1313–1323 (in Chinese with English abstract).
[11] 吕维娜. 花生栽培种SSR遗传连锁图谱构建及重要产量性状QTL定位分析. 郑州大学硕士学位论文, 河南郑州, 2014.
Lyu W N. Contruction of Genetic Linkage Map Based on SSR Markers and QTLs Identification for Major Yield Traits in the Cultivated Peanut (L.). MS Thesis of Zhengzhou University, Zhengzhou, Henan, China, 2014 (in Chinese with English abstract).
[12] 成良强. 花生遗传图谱构建及产量相关性状的QTL分析. 中国农业科学院硕士学位论文, 北京, 2014.
Cheng L Q. Construction of Genetic Linkage Map and QTL Analysis for Yield Related Traits in Peanut (L.). MS Thesis of Chinese Academy of Agricultural Sciences, Beijing, China, 2014 (in Chinese with English abstract).
[13] Shirasawa K, Koilkonda P, Aoki K, Hirakawa H, Tabata S, Watanabe M, Hasegawa M, Kiyoshima H, Suzuki S, Kuwata C, Naito Y, Kuboyama T, Nakaya A, Sasamoto S, Watanabe A, Kato M, Kawashima K, Kishida Y, Kohara M, Kurabayashi A, Chika T, Tsuruoka H, Wada T, Isobe S. In silico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut., 2012, 12: 80.
[14] 姜慧芳. 花生种质资源描述规范和数据标准, 3-9. 北京: 中国农业出版社, 2006. pp 355–455.
Jiang H F. Specification for Description and Data of Peanut Germplasm Resources, 3-9. Beijing: China Agriculture Press, 2016. pp 355–455 (in Chinese)
[15] 王亮, 杨鑫雷, Getahun A, 崔顺立, 穆国俊, 刘立峰, 李自超. 栽培种花生AFLP标记体系的优化及多态性引物筛选. 核农学报, 2017, 31: 2087–2095.
Wang L, Yang X L, Getahun A, Cui S L, Mu G J, Liu L F, Li Z C. Screening for polymorphic primer pairs and optimization of AFLP marker system in peanut., 2017, 31: 2087–2095 (in Chinese with English abstract).
[16] Lin Z X, He D H, Zhang X L, Nie Y C, Guo X P, Feng C D, Stewart J M. Linkage map construction and mapping QTL for cotton fibre quality using SRAP, SSR and RAPD., 2008, 124: 180–187.
[17] Yu J W, Yu S X, Lu C R, Wang W, Fan S L, Song M Z, Lin Z X, Zhang X L, Zhang J F. High-density linkage map of cultivated allotetraploid cotton based on SSR, TRAP, SRAP and AFLP markers., 2007, 49: 716–724.
[18] 崔顺立, 刘立峰, 陈焕英, 耿立格, 孟成生, 杨余. 河北省花生地方品种基于SSR标记的遗传多样性. 中国农业科学, 2009, 42: 3346–3353.
Cui S L, Liu L F, Chen H Y, Geng L G, Meng C S, Yang Y. Genetic diversity of peanut landraces in Hebei province revealed by SSR markers., 2009, 42: 3346–3353 (in Chinese with English abstract).
[19] JoinMap 4.0 Software for the Calculation of Genetic Linkage Maps in Experimental Populations. Wageningen: Kyazma B V, 2006.
[20] Kosambi D D. The estimation of map distances from recombination values., 2011, 1: 172–175.
[21] Voorrips RE. MapChart: software for the graphical presentation of linkage maps and QTLs., 2002, 93: 77–78.
[22] 陈建国, 朱军. 籼粳交稻米蛋白质含量的基因型与环境互作效应的分析. 作物学报, 1999, 25: 579–584.
Chen J G, Zhu J. Analysis of genotype by environment interaction for protein content incrosses of rice (L.)., 1999, 25: 579–584 (in Chinese with English abstract).
[23] 刘雪海, 田俊芹. 花生品种主要性状遗传力和遗传进度的估算. 华北农学报, 1981, 1(2): 48–50.
Liu X M, Tian J Q. Estimation of heritability and genetic progress of main traits of peanut varieties., 1981, 1(2): 48–50 (in Chinese).
[24] Meng L, Li H H, Zhang L Y, Wang J K. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations., 2015, 3: 269–283.
[25] 张志勇. 水稻粒型和粒重性状的主效QTL定位研究. 厦门大学硕士学位论文, 福建厦门, 2008.
Zhang Z Y. Mapping of Major QTL for Grain Shape and Weight Traits in Rice (L.). MS Thesis of Xiamen University, Xiamen, Fujian, China, 2008 (in Chinese with English abstract).
[26] Varshney R K, Bertioli D J, Moretzsohn M C, Vadez V, Krishnamurthy L, Aruna R, Nigam S N, Moss B J, Seetha K, Ravi K, He G, Knapp S J, Hoisington D A. The first SSR-based genetic linkage map for cultivated groundnut (L.)., 2009, 118: 729–739.
[27] Lu Q, Liu H, Hong Y B, Li H F, Liu H Y, Li X Y, Wen S J, Zhou G Y, Li S X, Chen X P, Liang X Q. Consensus map integration and QTL meta-analysis narrowed a locus for yield traits to 0.7 cM and refined a region for late leaf spot resistance traits to 0.38 cM on linkage group A05 in peanut (L.)., 2018, 19: 887.
[28] Liang Y B, Liang X Q, Chen X P, Zhou H Y, Li G Y, Shao X, Wen S J. Construction of genetic linkage map based on SSR markers in peanut (L.)., 2008, 7: 915–921.
[29] Hong Y B, Chen X P, Liang X Q, Liu H Y, Zhou G Y, Li S X, Wen S J, Holbrook C, Guo B Z. A SSR-based composite genetic linkage map for the cultivated peanut (L.) genome., 2010, 10: 17.
[30] Huang L, He H, Chen W G, Ren X P, Chen Y N, Zhou X J, Xia Y L, Wang X L, Jiang X J, Liao B S, Jiang H F. Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut (L.)., 2015, 128: 1103–1115
[31] 曾新颖, 郭建斌, 赵姣姣, 陈伟刚, 邱西克, 黄莉, 罗怀勇, 周晓静, 姜慧芳, 黄家权. 花生籽仁大小相关性状QTL定位. 作物学报, 2019, 45: 1200–1207.
Zeng X Y, Guo J B, Zhao J J, Chen W G, Qiu X K, Huang L, Luo H Y, Zhou X J, Jiang H F, Huang J Q. Identification of QTL related to seed size in peanut (L.)., 2019, 45: 1200–1207 (in Chinese with English abstract).
[32] Gomez S M, Narayana M, Schubert A M, Ayers J, Baring M R, Burow M D. Identification of QTLs for pod and kernel traits in cultivated peanut by bulked segregant analysis., 2009, 12: 1–10.
[33] Yuan A P, Cao L Y, Zhuang J Y, Li R Z, Zheng K L, Zhu J, Cheng S H. Analysis of additive and AE interaction effects of QTLs controlling plant height, heading date and panicle number in rice (L.)., 2003, 30: 899–906.
[34] Specht J E, Chase K, Macrander M, Graef G L, Chung J, Markwell J P, Germann M, Orf J H, Lark K G. Soybean response to water., 2001, 41: 493–509.
QTL mapping and QTL × Environment interaction analysis of pod and seed related traits in cultivated peanut (L.)
MENG Xin-Hao1, ZHANG Jing-Nan1, CUI Shun-Li1, Charles Y. Chen2, MU Guo-Jun1, HOU Ming-Yu1, YANG Xin-Lei1,*, and LIU Li-Feng1,*
1State Key Laboratory of North China for Crop Improvement and Regulation / Key laboratory of Crop Germplasm Resources of Hebei Province / Hebei Agricultural University, Baoding 071001, Hebei, China;2Department of Crop, Soil and Environmental Sciences, Auburn University, Auburn 36849, USA
Pod and seed traits are important agronomy traits, which are closely related to yield in cultivated peanut (L.). In the present study, to identify molecular markers closely linked to pod and seed traits, a RIL8population with 315 families was developed that derived from Jihua 5 with large pod and M130 with small pod of US germplasm. A genetic linkage map containing 363 polymorphic loci was constructed using SSR, AhTE, SRAP, and TRAP markers. All polymorphic loci were mapped on 21 linkage groups, which spanned 1360.38 cM with an average distance of 3.75 cM. Subsequently, a total of 97 QTLs for pod and seed traits were identified by ICIM method at five environments from 2017 to 2018, explaining the phenotypic variations of 2.36%–12.15% , and located on A02, A05, A08, A09, B02, B03, B04, B08, and B09 chromosomes. Among them, nine QTLs were detected for pod length, 13 QTLs for pod width, 14 QTLs for pod thickness, 11 QTLs for seed length, 13 QTLs for seed width, 13 QTLs for hundred-pod weight, 10 QTLs for hundred-seed weight. Four QTLs with major effect were detected, including,,,and, which explained the phenotypic variations of 10.02%–12.15%. Furthermore, 45 stable QTLs were repeatedly detected in more than three environments. QTL clusters were detected on A02, A08, B02, B04, and B08 chromosomes, respectively.In addition, 15 epistatic QTLs were identified that explaining phenotypic variation of 10.23%–51.84%. These results will provide an important theoretical basis for molecular marker-assisted breeding of pod and seed traits in peanut.
peanut; pod; seed; QTL; QTL×E
10.3724/SP.J.1006.2021.04216
本研究由国家现代农业产业技术体系建设专项(CARS-13), 国家自然科学基金项目(31701459, 31771833), 河北省科技计划项目(16226301D), 河北省现代农业产业技术体系油料创新团队项目(HBCT2018090202)和河北省青年拔尖人才项目(0602015)资助。
This study was supported by the Special Fund for Modern Agro-industry Technology Research System of China (CARS-13), the National Natural Science Foundation of China (31701459, 31771833), the Science and Technology Research and Development Program of Hebei Province (16226301D), the Earmarked Fund for Hebei Oil Crop Innovation Team of Modern Agro-industry Technology Research System (HBCT2018090202), and the Support Program for the Top Young Talents of Hebei Province (0602015).
杨鑫雷, E-mail: peanut@hebau.edu.cn; 刘立峰, E-mail: liulifeng@hebau.edu.cn
E-mail: mxinhao1994@126.com
2020-09-22;
2021-03-19;
2021-04-08.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20210407.1849.008.html
