棉花花器官突变体的鉴定及候选基因的克隆
2021-08-05杨琴莉杨多凤丁林云黄楚珺高梦涛严孙艺张天真
杨琴莉 杨多凤 丁林云 赵 汀 张 军 梅 欢 黄楚珺 高 阳 叶 莉 高梦涛 严孙艺 张天真, 胡 艳,,*
棉花花器官突变体的鉴定及候选基因的克隆
杨琴莉1杨多凤1丁林云1赵 汀2张 军2梅 欢2黄楚珺2高 阳1叶 莉1高梦涛1严孙艺2张天真1,2胡 艳1,2,*
1南京农业大学作物遗传与种质创新国家重点实验室, 江苏南京 210095;2浙江大学农业与生物技术学院, 浙江杭州 310058
棉花是世界性的重要经济作物, 是天然纤维的主要来源。棉花生殖生长过程现蕾、开花、结铃都直接影响棉花主要经济性状——棉纤维的产量和品质。本研究在转基因棉花材料中发现了1个花器官突变体, 命名为, 其花器官呈现瓣化特征。PCR和Southern杂交证明突变体中的外源基因已整合到基因组中, 且为单拷贝插入。通过基因组重测序进行序列比较发现, 突变体基因组中外源T-DNA插入位点为染色体A11:59086840。PCR和Southern杂交对插入位点进行了进一步验证。根据棉花基因组注释结果, 在基因组插入位点附近有3个候选基因(、和)。其中为类基因。已有研究证明,类基因为花器官ABC模型中控制萼片和花瓣形成的A类功能基因。qRT-PCR结果显示,在转基因受体W0的花瓣、雌蕊和雄蕊3个组织中的表达与突变体中存在显著性差异。本研究为进一步深入探究棉花花器官发育的分子机制研究提供了参考。
棉花; 转基因; 花器官; 突变体; 基因克隆
棉花是世界上重要的经济作物和油料作物, 在世界上超过100个国家和地区种植。棉花纤维是纺织工业天然纤维的重要来源[1]。纤维由棉花胚珠表皮细胞突起形成[2], 因此棉花花器官的分化和发育直接影响棉花的产量和纤维品质。在植株的生长发育过程中, 植物顶端分生组织由发育成叶原基转变为开始发育成花原基[3], 是营养生长到生殖生长的转变。花器官的分化和发育是被子植物生命周期的重要阶段[4]。典型的花器官分为4个部分, 从外到内依次是萼片、花瓣、雄蕊、心皮, 它们组成了花发育的4个轮回[5-6]。
1991年, Coen和Meyerowitz[7]首次系统地解释了花发育相关基因(主要是MADS盒基因)在不同花器官发育中的作用。已知MIKC型的MADS-box基因是植物特有的, 它们在开花植物(被子植物)生殖发育中的作用尤为重要[8]。绝大多数MIKC型基因属于花器官决定基因, 它建立了被子植物花发育的基本框架, 保障了“AE模型”[9]的保守性, 并参与调控开花时间、花器官和花分生组织识别、果实发育、营养器官发育[10-11]等过程。除了MADS-box基因家族外, 还有一类()基因在花发育过程中扮演了重要角色。基因属于花发育模型中A类基因。最早发现拟南芥胚珠和雌配子体发育所需的基因与花型同源异形基因有关。编码的蛋白质()属于相关基因家族, 是胚珠发育所必需的[12-13]。已有研究表明, 拟南芥中基因在其子房及雌配子体发育过程中发挥了关键作用[13-16]。同时为了更好地理解基因在控制胚珠发育中的作用, 研究人员从拟南芥中分离出1个雌性不育突变体。在该突变体的胚珠中, 胚珠不发育。当突变体与花同源突变体结合时, 花器官结构就会完全丧失, 表明这2个基因共同启动了花发育[12]。
有关棉花花器官发育相关基因已有报道。Wang等[17]从陆地棉中分别克隆到了1个类似基因和1个开花启动因子基因, 并在转基因拟南芥中进行了功能验证。Zhang等[18-19]对和基因家族进行了鉴定和分析, 证实与和基因启动子结合以调节开花。而后研究确定了和的功能及其调控机制, 为早期成熟棉花品种发展提供了有用的信息[19]。研究表明基因属于PI亚家族, 为B类功能基因, 基因属于亚家族, 行使C类基因功能[20]。烟草植物中导入和基因, 证实了基因在相应花器官发育中的作用[20]。此外,基因在植物的花发育网络调控中也扮演了重要角色。了解基因与其他基因的相互作用关系, 有利于我们更好地理解花发育调控的基本框架[21]。已知与同属于A类功能基因, 它们共同调控花萼和花瓣的发育, 且2个基因被证明有一定的叠加效应, 它们共同影响了花原基的建成[16]。在花的发育过程中,基因与基因作用最为明显。已有研究表明, 在突变体中, 很多负调控因子中的作用最强,将的表达限制在花器官形成的第1、2轮回中[22-23]。miR172对的表达也有一定的调控作用。由于结构中存在1个miR172的结合位点, 因此二者结合之后, miR172在翻译水平抑制了蛋白质的形成[21]。总之, 在各类基因相互作用下形成的复杂调控网络, 共同调节了植物的花发育进程。
本研究在转基因棉花中发现了1个花器官突变体。突变体表现为花器官异常, 萼片相对细长偏大, 花瓣和雄蕊结构异常。Southern杂交表明, T-DNA在基因组中为单拷贝插入。利用基因组重测序数据进行比较基因组分析, 鉴定出T-DNA在突变体基因组的插入位点进行了准确定位。插入位点附近存在3个候选基因, 其中包括1个类基因, 在突变花器官中表达量显著低于正常花器官。推测可能由于T-DNA的插入导致1个类基因的表达发生差异变化, 从而引起花器官突变表型。
1 材料与方法
1.1 试验材料
花器官突变体为转基因植株。植物材料种植于浙江大学农生环9号温室(培养条件: 温度22℃, 光照16 h/黑暗8 h, 相对湿度75%~80%)。试验中转基因载体为GoPGF-RNAi, 载体骨架序列为pBI121载体(图1)保存于本实验室。
1.2 DNA、RNA提取及cDNA合成
采用CTAB法提取转基因受体W0和花器官突变体新鲜叶片DNA。按照EASY spin Plus植物RNA快速提取试剂盒(Adlab, 北京)指示说明书, 分别提取W0和的萼片、花瓣、雄蕊和雌蕊4个组织的RNA。植物材料于液氮中速冻后保存于-80℃超低温冰箱中备用。使用反转录试剂盒将各组织RNA反转成cDNA。
1.3 Southern印记杂交
提取叶片DNA后, 取20 μg基因组DNA, 加入150 U的d III内切酶和20 μL限制性内切酶缓冲液, 加无菌重蒸水至总体积200 μL, 37℃保温过夜。d III内切酶(南京诺唯赞生物科技股份有限公司, 简称诺唯赞)酶切后的质粒作为阳性对照。以基因扩增产物(约196 bp)和T-DNA扩增产物做探针, 分别检测外源基因插入的拷贝数以及插入位点的验证。基因扩增引物序列为F: 5¢-ACCTGTCCGGT GCCCTGAATGAACTGC-3¢和R: 5¢-GCCATGATGGAT ACTTTCTCGGCAGGAGC-3¢; T-DNA插入片段扩增引物序列为F: 5¢-TGGGTGATGGTTCACGTAGT-3¢和R: 5¢-CATAATCATGTTAAAATGCTTGG-3¢。将酶切后的基因组DNA在0.8%琼脂糖凝胶中电泳过夜。通过纸吸印法转移到尼龙膜上, 参照DIG High Prime DNA Labeling and Detection Starter Kit I (Roche)试剂盒进行探针标记、预杂交、杂交及检测。
1.4 T-DNA插入位点鉴定
为明确外源T-DNA插入基因组的准确位点, 本研究对花器官突变体和受体W0进行高深度的Illumina PE150基因组重测序。对所获得数据进行过滤去除低质量reads。利用Blastn[24]软件将高质量reads比对pBI121载体序列。提取比对到pBI121载体序列的另一端数据, 使用软件hisat2[25]比对陆地棉TM-1参考基因组(V2.1) (cotton.zju.edu.cn), 估算插入位点。
1.5 候选基因的克隆
从本实验室TM-1基因组(V2.1)数据库中获取候选基因、和三个基因的CDS序列, 设计引物对3个基因进行克隆, 引物序列分别为(F: 5¢- ATGAAGTCCATGAGCAATGATG-3¢, R: 5¢-CTAAG CATCTGTCCAGGCAGT-3¢)、(F: 5¢- ATGCAAAAGGAGGCTGCTCTTTT-3¢, R: 5¢-CTATA GCTTACACATGAGC-3¢)、(F: 5¢-ATG GAAGGAAGTATACCCTT-3¢, R: 5¢-CTAGACAAAT TCATGTAAACCACG-3¢)。
1.6 实时荧光定量PCR (RT-PCR)
以棉花基因(Acc. No. AF024716, F: 5¢- GGTGGTGTGAAGAAGCCTCAT-3¢, R: 5¢-AATTTC ACGAACAAGCCTCTGGAA-3¢)为内参对表达数据进行标准化。使用诺唯赞SYBR Green I kit染料, 在荧光定量RT-PCR仪(ABI 7500)上进行分析, 以最小的样本阈值循环数(Ct值)和最高的荧光值为标准。反应体系与程序按照诺唯赞AceQ qPCR SYBR Green Master Mix试剂盒说明书进行。引物序列分别为:-QF: 5¢-ATGGAGGTTAGCAATC AAGG-3¢,-QR: 5¢-AGATGAGATGGT GGAGATGG-3¢;-QF: 5¢-GTTTAGAC GAGTGGAAGCC-3¢,-QR: 5¢-AGTTT GGAGTGTCCTAATGC-3¢;-QF: 5¢-G GCCTATAAAGCGTGTACC-3¢;-QR: 5¢-TAATTCGGAGCTACAAGTGC-3¢。采用2-ΔΔCt法计算相对表达量, 计算公式如下: 目的基因的相对表达量= 2-ΔΔCt; 而DCt = Ct目的基因-Ct内参。
2 结果与分析
2.1 花器官突变体182-9表型鉴定
本研究以棉花野生型W0为受体, 在转基因材料中发现1个转基因克隆株系表现为花器官突变体, 命名为。突变体花器官呈现明显的生理缺陷构造, 整个花器官显著变小, 外围苞叶相对卷曲窄小, 萼片相对细长偏大, 花瓣和雄蕊结构异常, 失去了花器官四轮结构, 均呈现类似细长叶片状态, 心皮中无胚珠(图2)。
2.2 突变体182-9的分子检测
对转基因突变体进行基因PCR检测发现,为转基因阳性克隆, 外源T-DNA片段已经整合到基因组中(图3)。
2.3 突变体182-9的Southern检测
为进一步确定T-DNA在突变体中的插入拷贝数, 本研究根据外源基因设计探针, 与转基因获得的不同克隆的基因组DNA (包括突变体)进行Southern杂交。同时, 以质粒DNA为阳性对照, 野生型W0为阴性对照。结果显示, 突变体中T-DNA已稳定整合到基因组中且为单拷贝插入(图4)。
A: 转基因受体W0花器官整体结构与子房。B: 突变体花器官结构。C: 转基因受体W0苞叶、萼片、花瓣、雌蕊雄蕊结构。D: 突变体苞叶、萼片以及瓣化结构。标尺为1 cm。
A: the whole flower organ and ovary of transgenic receptor W0. B: floral organ of the mutant. C: bract, sepal, petal, pistil stamen of transgenic receptor W0. D: bracts, sepals, and petaloid structure of the mutant. Bar: 1 cm.
2.4 插入位点在参考基因组中的定位
我们对转基因受体W0和突变体进行基因组重测序, 分别产生了70.8 Gb和73.7 Gb的数据, 覆盖基因组约30倍(以棉花基因组2.5 Gb计算) (表1)。其中, Sample代表样品名, Raw Base (bp)表示原始数据产量, Clean Base是过滤之后的有效数据量, 即过滤后测序序列的个数乘以测序序列的长度, Q30即Phred 数值大于30的碱基占总体碱基的百分比。为鉴定T-DNA在受体W0基因组中的插入位点, 本研究将重测序reads分别比对到载体pBI121, 在转基因材料和转基因受体W0重测序reads中, 分别有41条和0条序列和载体具有同源性。使用软件hisat 2[25]将41条reads的另一端再比对到TM-1的参考基因组上发现, 共有15条reads能唯一匹配到陆地棉TM-1 A11染色体的59,086,746~59,086,947的区间, 推测位点为外源T-DNA的插入位点。
M: DNA分子量Marker; W0: 阴性对照; 1: 转基因株系182-9; 2: 转基因株系182-36; 3: 转基因株系182-91; 4: 转基因株系182-150; 5: 转基因株系182-172; 6: 转基因株系182-173; 7: 转基因株系182-187。
M: DNA molecular-weight markers; W0: negative control; 1: transgenic line 182-9; 2: transgenic line 182-36; 3: transgenic line 182-91; 4: transgenic line 182-150; 5: transgenic line 182-172; 6: transgenic line 182-173; 7: transgenic line 182-187.
M: DNA marker; 1: 转基因受体W0; C: 质粒; 2: 转基因株系182-187; 3: 转基因株系182-36; 4: 转基因株系182-173; 5: 转基因株系182-9 (箭头所示)。
M: DNA marker; 1: transgenic receptor W0; C: plasmid; 2: transgenic line 182-187; 3: transgenic line 182-36; 4: transgenic line 182-173; 5: transgenic line 182-9 (indicated by the arrow).
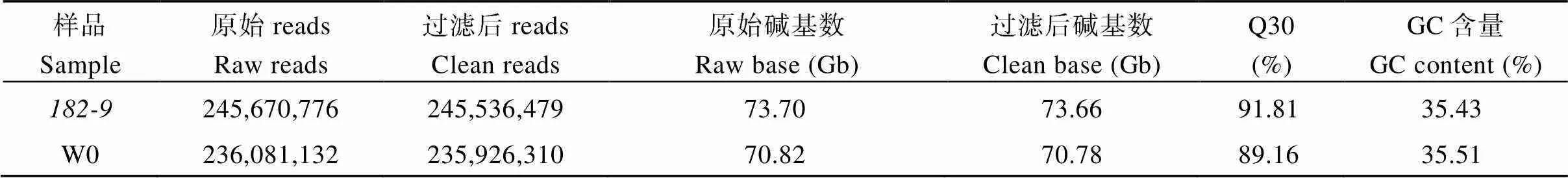
表1 重测序数据评估
为进一步验证外源T-DNA的插入位点。根据重测序分析结果, 本研究利用SeqHunter1.0提取陆地棉TM-1基因组中该位点上下游2 kb序列, 设计特异性引物, 分别以突变体和野生型W0的基因组DNA为模板, 扩增序列并测序。PCR扩增结果显示, 突变体能扩增出约400 bp的特异性序列(图5-A)。测序结果证明, 突变体基因组中T-DNA插入位点在染色体A11上的59,086,840位置, 红色部分为该位点附近的棉花基因组序列, 前半部分为载体序列(图5-B)。
A: PCR扩增条带; B: 扩增序列的测序结果(红色部分为棉花基因组序列, 其他为T-DNA序列)。M: DNA marker; W0: 转基因受体;: 转基因株系。
A: diagram of PCR detection; B: the sequencing results of the amplified sequence from(the red part is the cotton genome sequence, and the others are T-DNA sequences). M: DNA marker; W0: transgenic receptor;: transgenic line.
根据PCR验证结果, 本研究以图5-B中的序列为探针, 与转基因突变体基因组DNA进行Southern杂交。同时, 以探针序列为阳性对照, 转基因株系W0为阴性对照。结果进一步验证了基因组中外源T-DNA以单拷贝插入染色体A11上的59,086,840位点(图6)。
M: DNA marker; C: 阳性质粒; 1: 转基因株系; 2: 转基因受体W0。箭头所示为转基因中特异性条带。
M: DNA marker; C: positive plasmid; 1: transgenic line; 2: transgenic receptor W0. The specific band ofis shown by an arrow.
2.5 候选基因的筛选和功能预测
根据外源T-DNA插入基因组中的定位结果, 本研究将外源插入T-DNA序列比对到陆地棉TM-1基因组, 根据陆地棉TM-1参考基因组注释结果(表2), 在插入位点400 kb范围内存在、和三个功能基因(图7)。根据拟南芥中同源基因的功能注释发现,为类基因, 已有文献报道类基因在花器官分化中起着重要作用, 为开花模型中A类基因。
为验证是否因为候选基因本身序列的突变而产生突变表型, 本研究对3个候选基因在转基因受体W0和花器官突变体中的基因序列进行了比较发现, 3个候选基因CDS序列在和W0之间并无差异(图8~图10), 表明T-DNA插入并未造成3个候选基因的碱基序列差异。
选取W0和典型花器官结构进行定量分析。q-PCR分析结果显示,在突变体与转基因受体W0相应的花瓣(O2)、雄蕊(O3)和雌蕊(O4)中表现显著性差异, 在花瓣(O2)和雌蕊(O4)突变体表达量显著低于转基因受体。在2个材料中表达无显著差异,在转基因受体W0花器官和突变体中几乎不表达(图11)。
3 讨论
棉纤维为棉花胚珠表皮突起形成, 因此棉花的主要经济性状——棉纤维的品质和产量与棉花的生殖生长密切相关。但是, 目前关于棉花中花器官形成和发育的分子机制的研究报道并不多。本研究通过转基因得到花器官突变体, 是研究花器官发育的良好材料。本研究中, 我们鉴定出了准确的外源T-DNA插入位点, 并在插入位点附近发现了1个类基因家族基因。近年来, 关于基因直接或间接参与花发育的某一具体调控仍存在许多问题,对于其基因家族调节植物生长发育的方式及基因互作也不是十分清楚[21]。本研究中, 虽然T-DNA的插入位置与位置相差243 kb, 但是在DNA的三维空间上, 他们可能是靠近的。这种现象随着三维基因组研究[26]的发展, 已经有更多的证据证明了这种现象的存在。调控元件对基因的远程调控也已有报道。比如, 在玉米中, 已有报道证明一个控制光周期的基因转录起始位点上游57 kb的Harbinger转座元件CTOR的插入调控了该基因的表达[27]。这种远程调控的作用机制为我们关于候选基因与突变表型之间的可能关系提供了一些理论依据。因此, 我们推测在突变体中, T-DNA的插入可能引起了候选基因区域空间构象上的改变, 从而引起了基因的表达差异。突变体花突变产生与的关系还需要更进一步的试验去证明。
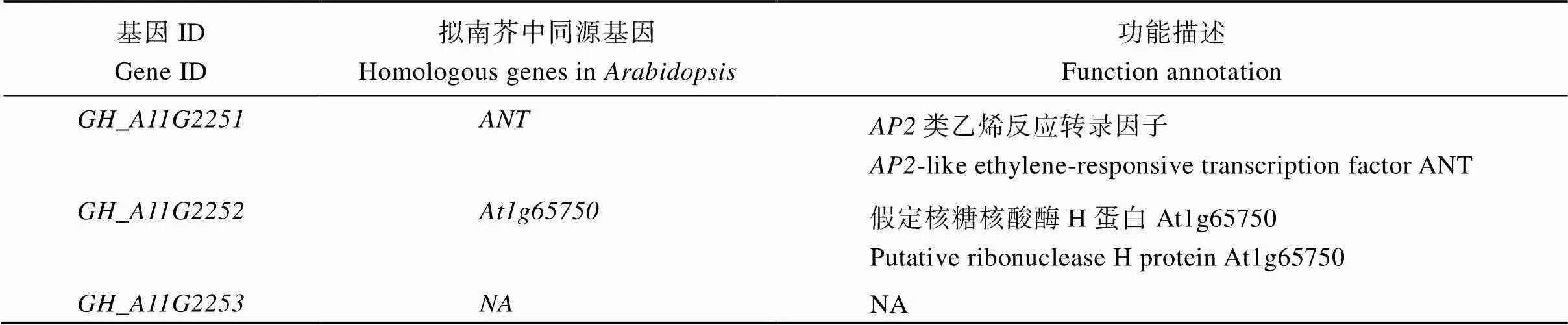
表2 候选基因的注释信息
4 结论
本研究通过花器官突变体与其野生型W0的表型差异, 为阐明调控棉花花发育的基因及突变机制提供了信息。鉴定出了T-DNA在转基因突变体中的准确插入位点A11:59,086,840, 并通过PCR以及Southern杂交进一步验证了位点鉴定的准确性。并在插入位点附近发现了一个可能性的功能基因, 在突变体与转基因受体W0的花器官中存在显著差异表达。
A: W0和组织示意图; B: 候选基因在突变体及其野生型W0中的表达水平。其中, *表示在0.05水平差异显著。
A: the schematic diagram of floral organs of W0 and; B: the expression levels of the candidate genes in mutantand wild-type W0. * meanssignificant difference at the 0.05 probability level.
[1] Zhang J, Guo W Z, Zhang T Z. Molecular linkage map of allotetraploid cotton (L. ×L.) with a haploid population., 2002, 105: 1166–1174.
[2] 陈良兵, 李永起. 棉花纤维发育的分子研究进展. 分子植物育种, 2004, 2: 105–110.
Chen L B, Li Y Q. The research progress on cotton fiber development at molecular level., 2004, 2: 105–110 (in Chinese with English abstract).
[3] Silva C S, Puranik S, Round A, Brennich M, Jourdain A, Parcy F, Hugouvieux V, Zubieta C. Evolution of the plant reproduction master regulators LFY and the MADS transcription factors: the role of protein structure in the evolutionary development of the flower., 2016, 6: 1193.
[4] Kotilainen M, Elomaa P, Uimari A, Albert V A, Yu D Y, Teeri T H.,an AGL2-like MADS box gene, participates in the C function during stamen development in., 2000, 12: 1893–1902.
[5] Theißen G. Development of floral organ identity: stories from the MADS house., 2001, 4: 75–85.
[6] 张云, 刘青林. 植物花发育的分子机理研究进展. 植物学通报, 2003, 20: 589–601.
Zhang Y, Liu Q Y. Proceedings on molecular mechanism of plant flower development., 2003, 20: 589–601 (in Chinese with English abstract).
[7] Coen E S, Meyerowitz E M. The war of the whorls: genetic interactions controlling flower development., 1991, 353: 31–37.
[8] Kaufmann K, Melzer R, Theißen G. MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants., 2005, 347: 183–198.
[9] 王亚杰. 巴西橡胶树MADS-box基因家族的克隆、表达谱分析及功能验证. 海南大学硕士学位论文, 海南海口, 2017.
Wang Y J. Molcular Cloning, Expression Profile and Functional Analysis of MADS-box Genes in. MS Thesis of Hainan University, Haikou, Hainan, China, 2017 (in Chinese with English abstract).
[10] Jiao Y, Wickett N J, Ayyampalayam S, Chanderbali A S, Landherr L, Ralph P E, Tomsho L P, Hu Y, Liang H, Soltis P S. Ancestral polyploidy in seed plants and angiosperms., 2011, 473: 97–100.
[11] Ng M, Yanofsky M F. Function and evolution of the plant mads-box gene family., 2001, 2: 186–195.
[12] Elliott R C, Betzner A S, Huttner E, Oakes M P, Tucker W Q J, Gerentes D, Perez P, Smyth D R. AINTEGUMENTA, an APETALA2-like gene ofwith pleiotropic roles in ovule development and floral organ growth., 1996, 8: 155–168.
[13] Klucher K M, Chow H, Reiser L, Fischer R L. Thegene of Arabidopsis required for ovule and female gametophyte development is related to the floral homeotic gene., 1996, 8: 137–153.
[14] Schmid M, Uhlenhaut N H, Godard F, Demar M, Bressan R, Weigel D, Lohmann J U. Dissection of floral induction pathways using global expression analysis., 2003, 130: 6001–6012.
[15] Aida M, Beis D, Heidstra R, Willemsen V, Blilou I, Galinha C, Nussaume L, Noh Y S, Amasino R, Scheres B. Thegenes mediate patterning of theroot stem cell niche., 2004, 119: 109–120.
[16] Zhao Q, Wang T, Wei X D. Function ofgene during floral organs development in higher plant: review., 2005, 259: 50–56.
[17] Wang X Y, Fan S L, Song M Z, Pang C Y, Wei H L, Yu J W, Ma Q F, Yu S X, Fang D D. Upland cotton geneconfers promotion of flowering time and shade-avoidance responses in, 2014, 9: e91869.
[18] Zhang X H, Dou L L, Pang C Y, Song M Z, Yu S X. Genomic organization, differential expression, and functional analysis of the SPL gene family in., 2015, 290: 115–126.
[19] Zhang X H, Wei J H, Fan S L, Song M Z, Pang C Y, Wei H L, Wang C S, Yu S Y. Functional characterization ofandhomologs from upland cotton (L.), 2016, 242: 178–186.
[20] 王力娜. 棉花MADS-box基因家族的克隆、表达谱分析及功能验证. 中国农业科学院硕士学位论文, 北京, 2010.
Wang L N. Molecular Cloning, Expression Profile and Functional Analysis of MADS-Box Genes in Cotton. MS Thesis of Chinese Academy of Agricultural Sciences, Beijing, China, 2010 (in Chinese with English abstract).
[21] 闻可心, 刘雪梅.功能基因在植物花发育中的重要作用. 生物技术通报, 2010, (2): 1–7.
Wen K X, Liu X M. The important role offunctional genes in plant floral developmen., 2010, (2): 1–7 (in Chinese with English abstract).
[22] Irish V F. Floral development in., 1998, 36: 61–68.
[23] Bomblies K, Dagenais N, Weigel D. Redundant enhancers mediate transcriptional repression of AGAMOUS by APETALA2., 1999, 216: 260–264.
[24] Altschul S F, Madden T L, Schffer A A, Zhang J H, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, 1997, 25: 3389–3402.
[25] Pertea M, Kim D, Pertea G M, Leek J T, Salzberg S L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown., 2016, 11: 1650.
[26] Lin D, Hong P, Zhang S H, Xu W Z, Jamal M, Yan K J, Lei Y Y, Li L, Ruan Y J, Fu Z F, Li G L, Cao G. Digestion-ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture., 2018, 50: 754–763.
[27] Huang C, Sun H Y, Xu D Y, Chen Q Y, Tian F. ZmCCT9 enhances maize adaptation to higher latitudes., 2018, 115: E334–E341.
Identification of a cotton flower organ mutantand cloning of candidate genes
YANG Qin-Li1, YANG Duo-Feng1, DING Lin-Yun1, ZHANG Ting2, ZHANG Jun2, MEI Huan2, HUANG Chu-Jun2, GAO Yang1, YE Li1, GAO Meng-Tao1, YAN Sun-Yi2, ZHANG Tian-Zhen1,2, and HU Yan1,2,*
1State Key Laboratory of Crop Genetics and Germplasm Enhancement, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China;2College of Agricultural & Biotechnology, Zhejiang University, Hangzhou 310000, Zhejiang, China
Cotton is an important economic crop and the main source of natural fiber in the world. The budding, flowering and bolling during cotton growth and development directly affect the yield and quality of cotton fiber that are the main economic traits of cotton. In this study, we found a flower organ mutant (named) in transgenic cottons, which displayed the floral organ petaloid feature. PCR and Southern blotting confirmed that the foreign T-DNA was integrated into thegenome with a single copy. Comparative analysis of the resequencing data revealed that the exogenic T-DNA was inserted in theon chromosome A11: 59086840. The insertion site was further verified by PCR and southern blot. According to the gene annotation of cotton genome, there were three candidate genes of,, and, near to the insertion site.encodedgenes controlling the formation of sepals and petals in the ABC model of flower organs as previous report. qRT-PCR showed that there were significant differences in the expression level ofin petals, pistil and stamens of transgenic receptor W0 and mutant. Our study provided the basis for further study of molecular mechanism in cotton floral organ development.
cotton; transgenic; floral organ; mutant; gene cloning
10.3724/SP.J.1006.2021.04208
本研究由国家自然科学基金项目(31970320)和国家转基因生物新品种培育重大专项(2016ZX08009-003)资助。
This study was supported by the National Natural Science Foundation of China (31970320) and the National Major Project for Developing New GM Crops (2016ZX08009-003).
胡艳, E-mail: njauhuyan@njau.edu.cn
E-mail: 739768392@qq.com
2020-09-10;
2021-01-13;
2021-03-11.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20210310.1713.002.html
