加权基因共表达网络分析筛胃癌相关分子
2021-07-29肖又德郑永法
肖又德 郑永法 戈 伟
1.泰康同济(武汉)医院肿瘤科,湖北武汉 430000;2.武汉大学人民医院肿瘤科,湖北武汉 430000
胃癌(gastric cancer)是世界上癌症相关性死亡的常见原因之一,其病死率位居癌症病死率的第三位[1],尽管以手术结合化疗的方案广泛应用于胃癌的治疗,但胃癌的5 年生存率依然较低[2-3]。基因芯片目前广泛应用于包括癌症在内的多种复杂疾病的诊断、预测以及药物筛选等领域[4-6]。但传统的筛选差异基因表达极易遗漏调控过程中的核心分子。通过系统的绘制个体生物网络互作图可以精准找出与预后相关的核心分子[7-8],加权基因共表达网络分析(weighted gene coexpression network analysis,WGCNA)可以有效解决上述问题[9-10]。本研究拟利用该方法筛选更为有效的胃癌分子标志物。
1 资料与方法
1.1 数据集获取
从TCGA 数据库(https://portal.gdc.cancer.gov/,检索时间:建库至2020 年11 月14 日)中下载胃癌患者的RNA-seq 数据及其临床资料,并剔除生存情况及病理分期不明的患者,最终纳入359 例胃癌患者。同时 从GEO 数 据 库(https://www.ncbi.nlm.nih.gov/geo/)中,通过关键词(胃癌、生存和基因芯片)检索筛选带有生存数据的胃腺癌数据集,且数据集能获得标准化的注释文件,获取两个数据集(GES15459 和GSE22377,检索时间:建库至2020 年11 月14 日),用于外部验证胃癌患者的预后。含有多种癌症组织和癌 旁 组 织 的ONCOMINE 数 据 库(https://www.oncomine.org)也用于外部验证。
1.2 基因共表达网络构建
WGCNA 是一种常用的模块化分析技术,已被用于识别和筛选复杂疾病的生物标志物或药物靶点[11]。首先,通过R 软件中的“WGCNA”分析包构建基因共表达网络[8,12]。然后,建立两个基因之间的相关矩阵,通过对网络拓扑结构分析,确定软阈值大小,将邻接转化为拓扑重叠矩阵(TOM),TOM 可以度量1个基因的网络连通性[7,13]。为了将表达谱相似的基因分类到基因模块(ME)中,基于TOM 的差异测度进行平均连锁层次聚类[8,12]。模块鉴定后,采用t 检验计算组间各基因表达显著性检验的P 值。为进一步分析模块特征基因的差异性,为模块树状图选择一条切线,并合并部分模块。
1.3 特征模块和核心基因筛选
对每个ME 进行分析时,通过Pearson 相关检验来评估MEs 与转移的相关性,以确定相关模块(各模块在各个样本上Pearson 系数绝对值相加为最高者认定为响应系数最高模块)。选择与转移高度相关的模块作为转移模块进行分析。利用STRING 网站进行蛋白-蛋白的互作(PPI)分析(选择连接数最大10个),并利用Cytoscape 软件中的MCODE 插件进一步分析。同时利用R 软件的LASSO 回归分析包筛选转移模块中基因。
1.4 GO 功能学、KEGG 通路分析和GSEA 分析
为了解转移模块涉及的功能学和通路。采用标准富集计算方法进行GO 功能分析和KEGG 通路分析用以筛选与其相关的功能和通路。为了解核心分子在胃癌患者中的潜在功能,利用TCGA 胃癌患者数据集进行GSEA 分析。将P<0.05 和|富集评分(ES)|>0.3作为截止标准。
1.5 统计学方法
采 用SPSS 24.0、GraphPad Prism 7.0 和R 3.4.1软件进行数据分析,同时用上述软件进行图像生成处理。组间比较采用t 检验。基因最佳截点取值,利用X-Tile 软件,基于最小P 值和最大Log-rank χ2值进行分析计算[14]。绘制受试者工作特征(ROC)曲线,评估核心靶基因的预测能力,利用曲线下面积(AUC)评估敏感度和特异性。K-M 法绘制生存曲线,预测基因对患者预后的影响。以P <0.05 为差异有统计学意义。
2 结果
2.1 患者一般资料
本研究共纳入359 例胃癌患者进行分析,其中男230 例,女129 例;年龄(65.7±10.9)岁;肿瘤位置:近端87 例,胃体134 例,远端131 例,未知7 例;肿瘤组织学分级:G18 例,G2128 例,G3215 例,GX8 例;病理分期:Ⅰ期53 例,Ⅱ期116 例,Ⅲ期152 例,Ⅳ期38 例。
2.2 构建共表达模块结果
WGCNA 方法将基因进行模块化富集分析(图1,封三),共筛选16个相应的ME,见图2(封三),依据各模块在各个样本上Pearson 系数筛选获取了BLUE模块,BLUE 模块共含有2078个基因。

图1 基因共表达网络分析模式图
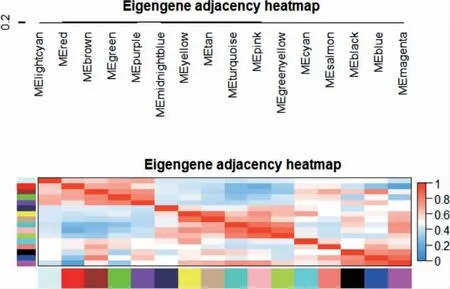
图2 各模块相关分析图
2.3 BLUE 模块相关功能分析及核心分子筛选结果
BLUE 模块中相关基因筛选表达响应基因参与的信号通路关联提示,主要与蛋白磷酸化异常、蛋白激酶活化等相关,见图3(封三)。PPI 和MOCDE 插件筛选位于调控核心位置的基因,见图4。LASSO 分析筛选出19个与预后相关的靶基因(DUSP16、NUP50、CDC5L、PTPRF、KBTBD2、SLC11A2、DHX15、ACBD5、RMI1、TRIM25、C3orf38、MSI2、VPS35、RNF43、PACRGL、GMCL1、SMAD5、IMPACT、IDE)。将上述3种方法筛选的结果进行综合分析,获得CDC5L。
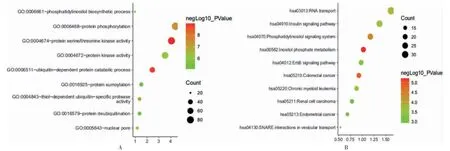
图3 BLUE 模块中GO 和KEGG 分析
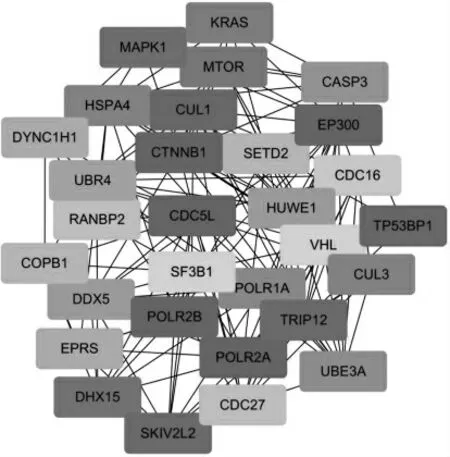
图4 蛋白互作分析
2.4 CDC5L 验证结果
CDC5L 表达量为7.2 时为最佳截点,见图5(封三)。低表达CDC5L 患者的总生存率高于高表达患者(P=0.038),见图6。为了解CDC5L 表达量在患者TNM 分期中的作用,对其分层分析,结果提示其Ⅰ、Ⅱ期低表达CDC5L 患者总生存率高于高表达患者(P=0.06、0.03),见图7。CDC5L 显示出较强的预测胃癌转移患者能力(AUC=0.745),见图8。ONCOMINE数据库对CDC5L 基因进行meta 分析发现,纳入的33个数据集中,癌组织中CDC5L 表达量高于癌旁组织(P <0.001),见图9。GEO 数据库验证结果显示,低表达CDC5L 的胃癌患者有着更好的预后,见图10。

图5 X-tile软件选取得CDC5L最佳截点
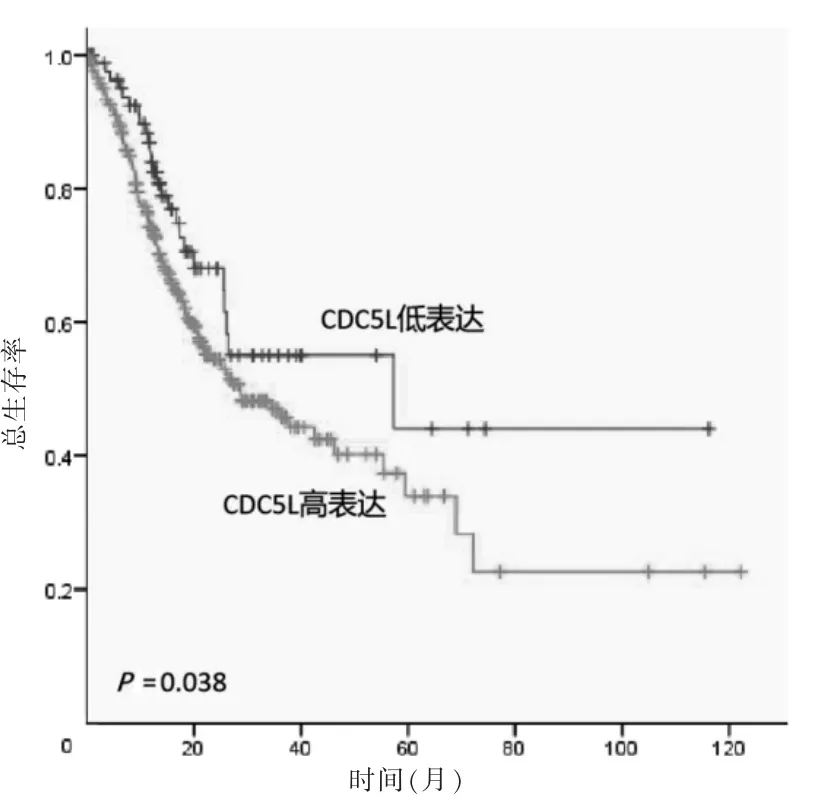
图6 不同CDC5L 表达情况胃癌患者生存分析曲线

图7 不同病理分期胃癌患者生存分析曲线
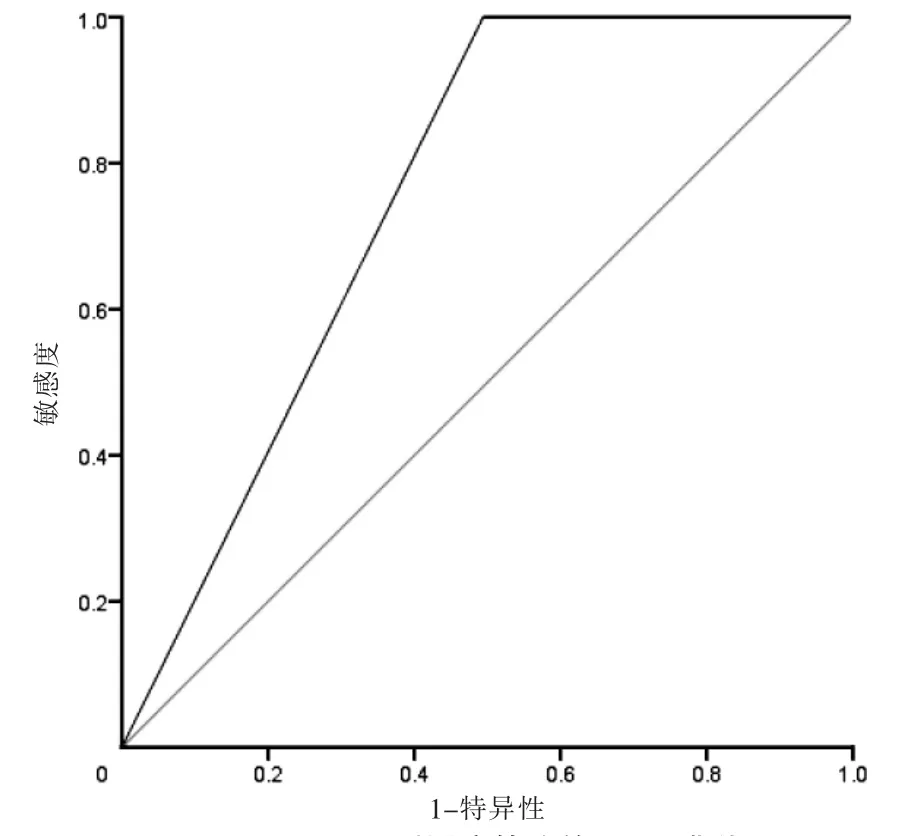
图8 CDC5L 预测胃癌转移的ROC 曲线

图9 胃癌和癌旁组织中CDC5L 表达比较
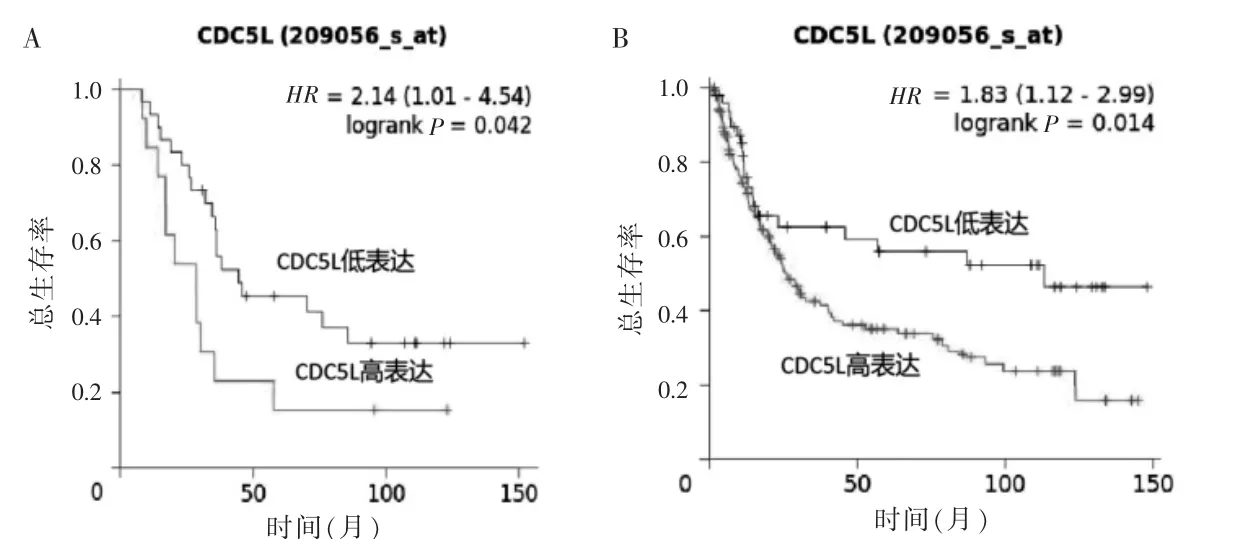
图10 CDC5L 在GEO 数据集的验证结果
2.5 GSEA 功能学分析结果
结果显示,CDC5L 主要涉及细胞周期调控以及糖代谢通路。见图11。
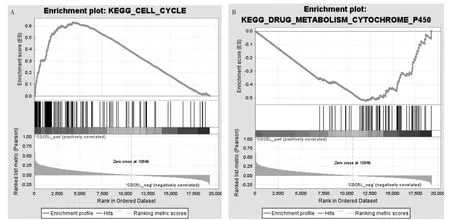
图11 GSEA 分析功能学分析结果
3 讨论
胃癌是世界上癌症相关性死亡的常见原因之一,尽管目前以手术结合化疗/免疫治疗/靶向治疗等多种方案已广泛应用于胃癌的治疗当中,但胃癌的五年生存率依然低下[2-3]。这主要是因为大多数胃癌患者被发现时已处于中晚期,造成其预后不佳[15]。晚期转移性胃癌患者五年生存率不足10%[16-17]。为更早地识别具有高危转移风险的胃癌患者,本研究利用TCGA 数据集,通过精准的WGCNA 方法识别出CDC5L 与胃癌患者预后相关,同时其能够较好地预测早期胃癌患者是否发生转移;内部和外部数据集同时验证CDC5L 具有较强的重复性和可靠性。
CDC5L 是G2/M 转变所必需的细胞周期调节剂[18-20]。CDC5L 通过与细胞周期检查点蛋白ATR 相互作用,激活ATR 下游效应因子,干扰CDC5L 表达使S期细胞周期检查点失活,从而增加药物敏感性[21-22]。研究显示[23-25],CDC5L 在肝癌、口腔鳞癌和神经母细胞瘤中也发挥了作用。但目前尚无CDC5L 在胃癌中的作用及相关机制的报道。本研究结果显示,CDC5L 能较好地预测胃癌患者生存及转移发生情况。外部ONCOMINE 数据库发现,CDC5L 在癌组织中高表达。GEO 数据集验证了低表达CDC5L 胃癌患者有更好的预后。因此,本研究结果对未来探索CDC5L 与胃癌患者不良预后奠定了一定的基础。但不可否认的是,本研究结果还需进一步在临床样本中获得更为可靠的验证,以及通过体内/外实验验证CDC5L 的临床和生物学意义。
总之,本研究通过WGCNA 等生物信息学方法利用TCGA 数据库发现CDC5L 高表达与较差预后相关。利用外部数据集对上述结果验证,发现CDC5L 在癌组织中高表达。CDC5L 有可能成为新的预后指标,有助于胃癌患者个性化治疗及临床预后判断。
