2型糖尿病治疗药物托格列净的合成路线图解
2021-07-29胡家栋文雯宋慧颖李博姚宇
胡家栋,文雯,宋慧颖,李博,姚宇
(1.杨凌职业技术学院药物与化工学院,陕西杨凌 712100;2.西北农林科技大学化学与药学院,陕西 杨凌 712100)
据世界卫生组织(WHO)和美国糖尿病协会(ADA)报道,糖尿病被分为1型糖尿病(T1DM)、2型糖尿病(T2DM)、妊娠期糖尿病和其他糖尿病4种类型。其中,2型糖尿病临床占比达90%以上,是最常见的类型[1‑4],其特征是胰岛素分泌不足和胰岛素抵抗导致的糖、脂肪和蛋白质代谢失调。目前,按照药物的作用机制和结构类型将糖尿病临床治疗药物分为以下7类[5]:①促胰岛素分泌剂(磺脲类和格列奈类);②双胍类;③噻唑烷二酮类;④α葡萄糖苷酶抑制剂(AGI);⑤胰岛素及其类似物;⑥胰高血糖素样肽1受体激动剂(GLP‑1 RA)和二肽基肽酶‑4(DPP‑4)抑制剂;⑦钠‑葡萄糖协同转运蛋白2抑制剂(SGLT‑2)。
钠‑葡萄糖协同转运蛋白(SGLT)是一类在小肠黏膜和肾脏近曲小管中发现的转运基因家族,肾脏对葡萄糖的重吸收主要依赖于SGLT蛋白介导。其中,位于近端肾小管S1段的SGLT‑2负责肾脏中约90%的葡萄糖重吸收。根皮苷衍生的SGLT‑2抑制剂竞争性地抑制了SGLT‑2蛋白的活性[6],减少了肾脏对葡萄糖的重吸收,增加尿糖的排出以降低血糖含量。该类药物的优点在于降糖机制不依赖胰岛素[7]。
截至目前已上市的SGLT‑2抑制剂有7种[8‑11],包括卡格列净(强生)、达格列净(百时美施贵宝&阿斯利康)、依帕列净(勃林格殷格翰&礼来)、伊格列净(安斯泰来)、托格列净(中外制药&赛诺菲)、鲁格列净(大正制药)和埃格列净(默克&辉瑞)。其中,口服托格列净(Tofogliflozin,1)于2014年在日本获批准上市,用于治疗成人2型糖尿病。
从结构上看,托格列净属于C‑芳基糖苷类化合物,其芳基苷元通过C‑C键与葡萄糖相连,芳环糖苷键邻位与糖基氧环合构成呋喃环。截至目前,文献报道的托格列净的合成路线有5种[12‑20],从策略上看(图1),可以分为两类:一类是通过亲核加成/直接环合法构建螺缩酮结构;另一类是通过分子内[4+2]环加成反应构建螺缩酮结构。
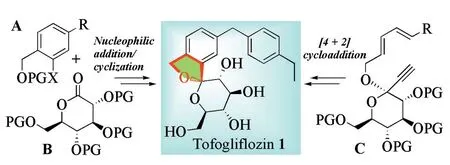
图1 托格列净(1)的合成策略Figure 1 Synthesis strategy of Tofogliflozin(1)
随着格列净类逐步被纳入医保[21]及其优秀的临床表现[22],该类药物市场前景广阔。本文从2个不同合成策略的角度对报道的托格列净合成路线进行综述,以期为其原料药的工业生产提供参考。
1 直接环合法构建螺缩酮结构
1.1 先构建C‑芳基糖苷键再形成二芳基糖苷配基
在从根皮苷(Phlorizin)类似物中寻找具有降血糖活性的先导化合物过程中,研究人员发现O‑芳基糖苷类化合物大多在体内实验中表现出代谢不稳定性,易被相应的糖苷酶水解。因此,结合计算机辅助药物设计(CADD)构建了一系列C‑芳基糖苷类化合物[13]。托格列净1就是其中之一,其首条合成路线[12‑13]模仿了抗真菌抗生素Papulacandins A‑D的合成策略[23]构建关键的C‑芳基糖苷键及其螺缩酮结构。
该路线由Kobayashi等[12‑13]提出(图2)。从商业可得的溴代对苯二甲酸2出发,经硼烷还原羧基得溴代对苯二甲醇3,醇羟基用三苯甲基(Tr)保护得取代溴苯4。接着,仲丁基锂与4在甲苯中发生锂‑卤交换形成芳基锂试剂对苄基保护的D‑葡萄糖酸δ‑内酯5加成构建C‑芳基糖苷键形成中间体6。糖苷6在三氟化硼乙醚和三乙基硅氢条件下脱Tr保护并环合构建螺缩酮结构形成了中间体7。螺缩酮7中苄醇经Dess‑Martin氧化得苯甲醛8,用格氏试剂9或者锂‑卤交换后的锂试剂10对醛基加成构建二芳基糖苷配基形成化合物11,二芳基碳桥上的仲醇被三乙基硅氢还原得苄基保护的目标产物12。最后,由氢气/Pd(OH)2‑C的条件脱除Bn保护以8步6.5%的总收率获得托格列净1。
该路线是托格列净实验室药物开发研究的第1条路线,尚存在以下几个问题:①总收率不高,不能大量合成而无法满足进一步的体内活性评价;②合成过程中缺乏可结晶的中间体,在多步反应中均需要利用费时费力的柱层析手段对中间体及终产物进行纯化。
基于路线一(图2)的局限性,Murakata等[14‑15]报道了第2条合成路线(图3)。从2,4‑二溴苄醇13出发,醇羟基经2‑甲氧基丙烯保护得中间体14。通过优化的锂‑卤交换条件[15],在14的甲苯‑甲基叔丁基醚溶液中加入0.8当量正丁基锂的正己烷溶液并在−10℃搅拌30 min后,继续加入0.3当量的正丁基锂以驱动反应完成。14保护基上的甲氧基与锂试剂的配位效应控制了锂‑卤交换的区域选择性,以53∶1的邻位选择性(98%的转化率)获得锂试剂15。接着,将芳基锂盐15加入到TMS保护的D‑葡萄糖酸δ‑内酯16中形成C‑芳基糖苷键;再加入TMSCl捕获糖苷键位的游离羟基得芳基糖苷17。再进行第二次锂‑卤交换,并加入对乙基苯甲醛18构建二芳基糖苷配基。之后,在19的四氢呋喃‑水溶液中加入1 mol/L的盐酸一锅法[24]脱TMS和2‑甲氧基丙烯保护并环合构建螺缩酮结构得到了羟基托格列净20。在乙二醇二甲醚中催化氢解20得到托格列净粗品1。
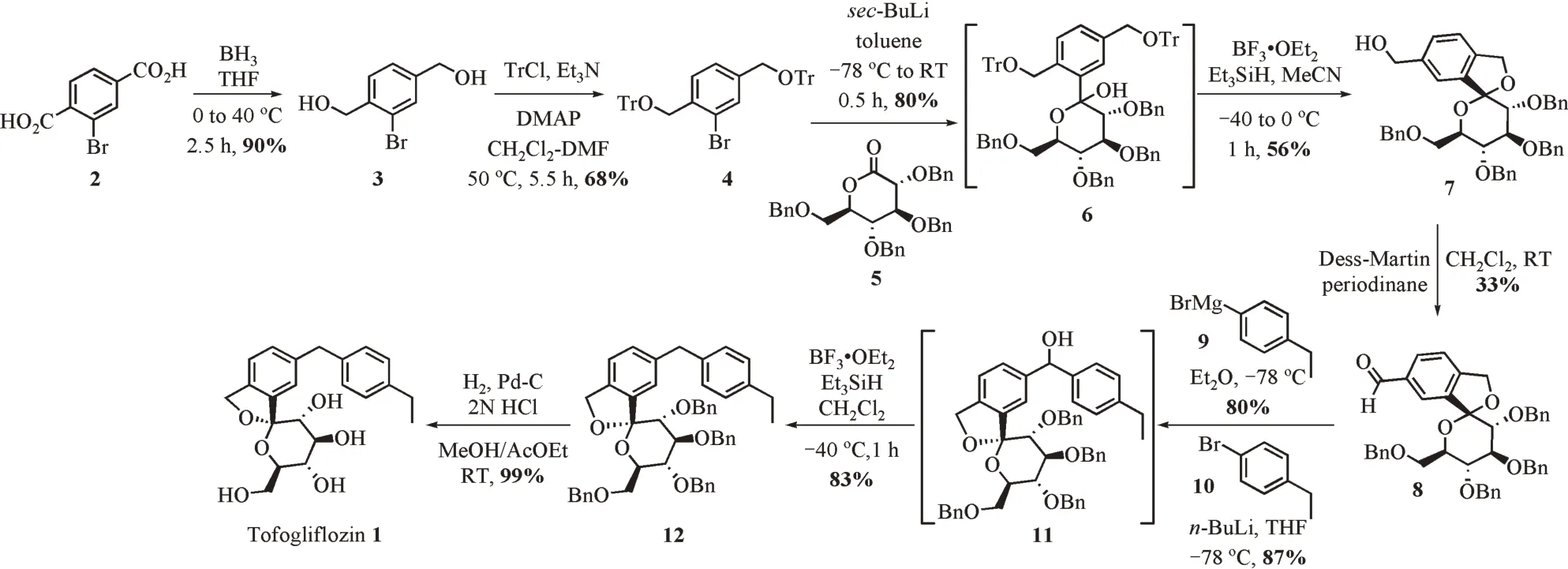
图2 文献[12‑13]报道的合成路线一Figure 2 The first synthetic route described in literatures 12&13
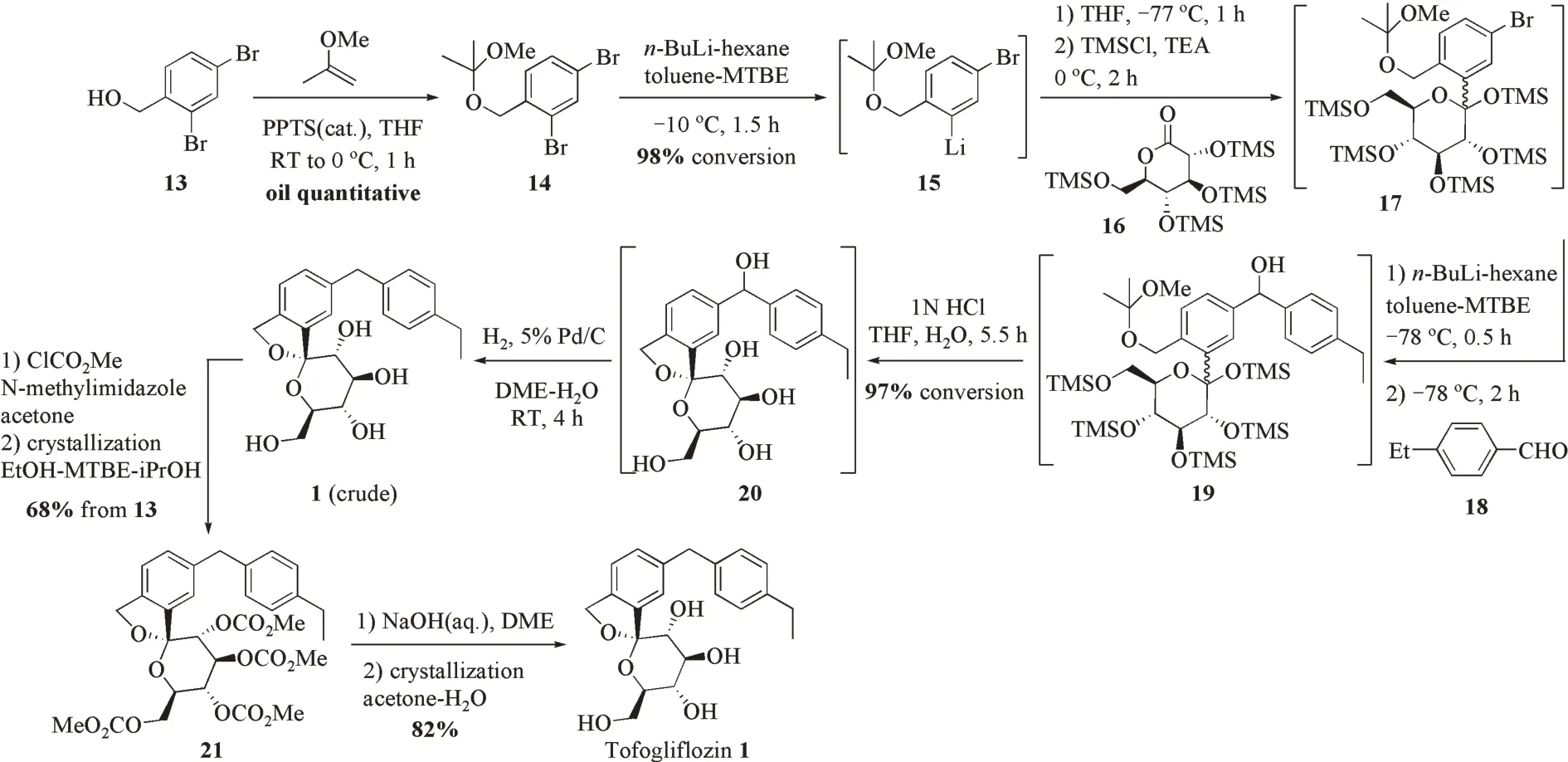
图3 文献[14‑15]报道的合成路线二Figure 3 The second synthetic route described in literatures 14&15
在制备了1的粗品之后,将糖基上的羟基用甲氧羰基保护得碳酸酯21。将中间体21在乙醇‑甲基叔丁基醚‑异丙醇体系中重结晶后得纯化的碳酸酯21。最后,再用NaOH脱保护,在丙酮‑水体系中重结晶后得到精制后的托格列净1。该路线共8步反应,总收率从路线一的6.5%提高至47%。
与合成路线一(图2)相比,该路线显著提高了API的总收率,但在精制方面,由于过程中没有合适的固体产物可以结晶纯化,因而需将粗产品进一步碳酸酯化后再进行结晶精制,增加了纯化步骤,尚有继续优化的空间。
为了进一步简化工业流程,缩短操作步骤,Ohtake等[18]将路线二中重结晶纯化的碳酸酯中间体21设计到了合成路线当中,在合成终产物之前就可以完成重结晶精制过程。该路线简化了路线二中将粗产品碳酸酯化再结晶精制的过程,将步骤缩短至6步,以39%的总收率完成了托格列净1的合成(图4)。
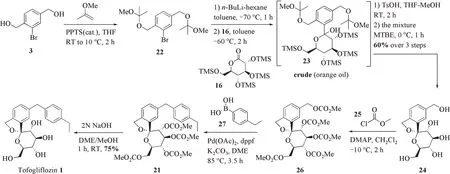
图4 文献[18]报道的合成路线三Figure 4 The third synthetic route described in literature 18
从溴代对苯二甲醇3出发,经2‑甲氧基丙烯保护二醇羟基得取代溴苯22,在中间体22的甲苯溶液中加入正丁基锂形成芳基锂盐中间体,以其对TMS保护的D‑葡萄糖酸δ‑内酯16加成构建C‑芳基糖苷键得到中间体23。芳基糖苷23在对甲苯磺酸‑四氢呋喃‑甲醇的条件下,一锅法脱TMS和2‑甲氧基丙烯保护并环合构建螺缩酮结构得到多羟基化合物24。粗品在甲基叔丁基醚中研磨得到结晶的中间体24时进行第1次纯化。接着,在氯甲酸酯‑DMAP条件下对化合物24的所有醇羟基用甲氧羰基保护得碳酸酯25。苄醇的羟基被甲氧羰基保护后可以利用Kuwano及其同事[25]的方法,与对乙基苯硼酸27发生Suzuki‑Miyaura偶联反应构建二芳基糖苷配基形成可结晶的碳酸酯中间体21。该步反应的Pd残留可以通过加入N‑乙酰‑L‑半胱氨酸(NAC)捕获除去,经过后处理可以获得纯度99%和小于10−6钯残留的21晶体。最后,21经NaOH水解获得托格列净1。
该路线在制备过程中不使用柱层析的纯化手段,总收率较好,可以用于托格列净的规模化制备。
1.2 先构建二芳基糖苷配基再形成C‑芳基糖苷键
前述3条路线都是先构建C‑芳基糖苷键并构筑螺缩酮环之后再完成二芳基糖苷配基的连接,Yang及其同事[19]报道了一种先通过Friedel‑Crafts酰基化反应构建二芳基糖苷配基,再形成C‑芳基糖苷键的路线(图5)。
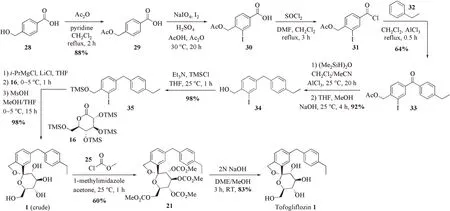
图5 文献[19]报道的合成路线四Figure 5 The fourth synthetic route described in literature 19
从商业可得的4‑羟甲基苯甲酸28出发,用乙酰基保护羟基得取代苯甲酸29。然后,在高碘酸钠‑碘单质的条件下经Lulinski优化的条件[26]选择性地在羧基间位引入碘原子。所得取代碘苯30在氯化亚砜和催化量DMF条件下制备苯甲酰氯31。化合物31在AlCl3催化下与乙基苯32发生Friedel‑Crafts酰基化反应构建二芳基配基33。接着,优化Kiuchi还原羰基的方法[27],在AlCl3为酸、1,1,3,3‑四甲基二硅氧烷为还原剂的条件下还原羰基为亚甲基。由于乙酰基保护基与后期形成C‑芳基糖苷键所用到的有机金属试剂不兼容,所以在这一步将Ac换成TMS保护基。先用NaOH脱乙酰基保护得二芳基碘苯34,再用TMS保护其醇羟基得二芳基糖苷配基35。用i‑PrMgCl·LiCl在−20℃条件下处理碘苯35形成有机金属试剂,再对TMS保护的D‑葡萄糖酸δ‑内酯16加成构建C‑芳基糖苷键;用对甲苯磺酸脱TMS保护并环合构建螺缩酮结构得到托格列净1的粗品。最后,采用和路线二相同的精制策略获得精制的托格列净1。
本条路线用12步的线性步骤,23%的总收率完成最终API的百克级合成。虽然路线较长,但因全程不使用昂贵的过渡金属催化剂,克服了重金属残留问题且降低了生产成本,仍具有一定的优势。
2 环加成法构建螺缩酮结构
上述4条路线均是通过芳基金属试剂对保护的D‑葡萄糖酸δ‑内酯的亲核加成,C‑芳基糖苷键处生成的羟基再与芳基上的苄醇直接环合而构建螺缩酮结构的。Kawase等[20]报道了一种通过分子内[4+2]环加成反应一步构建螺缩酮结构的策略(图6)。
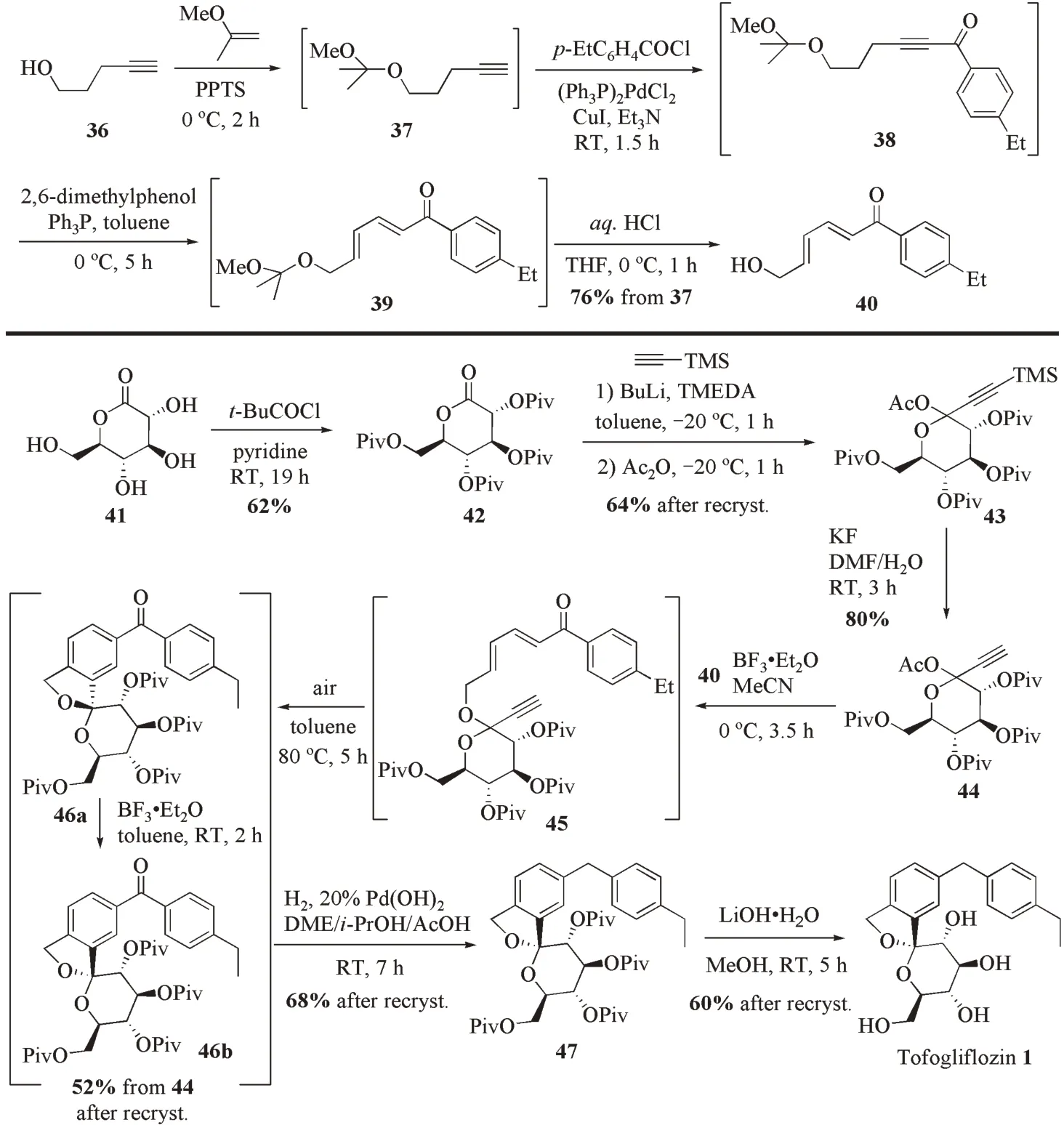
图6 文献[20]报道的合成路线五Figure 6 The fifth synthetic route described in literature 20
路线从商业可得的4‑戊炔‑1‑醇36出发,经2‑甲氧基丙烯保护醇羟基得中间体37,戊炔37在三苯基膦氯化钯‑碘化亚铜‑三乙胺催化下和对乙基苯甲酰氯偶联得炔酮38。中间体38在2,6‑二甲基苯酚和三苯基膦条件下[28]异构化为二烯酮39,进一步在稀盐酸条件下脱保护得双烯体片段40。在完成了双烯体片段的构建后,再从D‑葡萄糖酸δ‑内酯41出发,经特戊酰基保护所有羟基得42。经Piv保护的葡萄糖酸内酯42被TMS保护的乙炔锂试剂加成后,形成的醇锂盐被醋酸酐捕获得到中间体43,其在氟化钾作用下脱去炔基的TMS保护得到含炔基的亲双烯体片段44。然后,双烯体片段40和炔基片段44在三氟化硼乙醚条件下发生亲核取代反应生成环加成的前体45。关键中间体45在甲苯80℃加热的条件下发生[4+2]环加成反应,随后在空气中发生氧化芳构化反应形成二氢异苯并呋喃骨架而构建了螺缩酮结构,生成的是46的一对非对映异构体的混合物。其中,46a可以在三氟化硼乙醚作用下异构化为构型正确的46b。中间体46二芳基桥的羰基在氢气‑Pd(OH)2条件下发生还原‑氢解反应生成亚甲基桥。最后,LiOH脱去特戊酰基保护得到终产物托格列净1。
该策略以12步5%的总收率完成了目标API的合成。尽管该路线与前4条路线相比,在步骤和收率上还有优化的空间,但也为该类药物的工业合成提供了新的思路。并且,这种方法还合成了托格列净1在C‑芳基糖苷键位置的差向异构体,这对托格列净杂质分析方法的建立具有重要意义。
3 总结与展望
合成托格列净1的关键在于含C‑芳基糖苷键的螺缩酮结构的构建,目前报道的5条合成路线主要通过2种策略来构建该结构:一种是芳基苷元的有机金属试剂对保护的D‑葡萄糖酸δ‑内酯亲核加成,原位生成的羟基再与芳环上的苄醇部分发生环合构建;一种是制备含双烯体/亲双烯体的糖苷后发生分子内[4+2]环加成再氧化芳构化构建。前4条路线属于策略一的方案,路线一是托格列净初期研发的药物化学合成路线,过程中无可结晶的中间体且总收率较低,路线二将托格列净粗品进行碳酸酯化后得到可结晶中间体精制后以8步47%的总收率制备了API,路线三中有2步可以结晶的中间体且将路线二中的碳酸酯中间体提前至合成过程中而缩减了合成步骤,路线四先完成二芳基侧链的构建且过程中不使用昂贵的过渡金属催化剂,降低了生产成本。路线五属于策略二的方案,为托格列净螺缩酮结构的构建提供了新的思路且完成了C‑芳基糖苷键位置差向异构体的合成,对质量控制中的杂质分析具有实践意义。目前,托格列净作为高选择性SGLT‑2抑制剂已经在日本上市使用,随着四期临床数据的进一步收集,托格列净的市场有望持续增长,未来可能在国内上市。本文通过系统总结托格列净的合成路线,期望为其后期的原料药工业生产提供参考。
