HPLC指纹图谱结合化学计量学评价不同规格野菊花药材质量
2021-07-29施文婷莫秋怡彭帮贵胡懿钟霞邓桂海王寿富
施文婷,莫秋怡,彭帮贵,胡懿,钟霞,邓桂海,王寿富
(广东一方制药有限公司/广东省中药配方颗粒企业重点实验室,广东佛山 528244)
野菊花是菊科植物野菊Chrysanthemum indi-cumL.的干燥头状花序,秋冬两季花初开时采摘,呈类球形,直径0.3~1.0 cm。野菊花味苦、辛,性寒,可清热解毒、泻火平肝,用于治疗疔疮痈肿、目赤肿痛、头痛眩晕[1]。野菊花广泛分布于我国的东北、华北、西北、华东和西南等地[2]。现代研究表明,野菊花的主要药效成分为黄酮类、酚酸类、多糖类和挥发油类物质[3‑6],主要药理作用包括抗菌、抗病毒、抗感染、免疫、神经保护、保肝和抗肿瘤等,对心血管疾病的防治和辅助抗血小板凝聚也有一定作用[7‑13]。
中药材的质量优劣与其内在的药效成分含量高低密切相关[14],张丽先等[15]通过建立HPLC指纹图谱对不同产地的野菊花药材进行了质量比较;郑继标等[16]对野菊花药材不同部位的蒙花苷含量进行了比较;魏民等[17]对野菊花药材在不同采收期的蒙花苷含量进行了研究,尚未见不同规格野菊花药材质量比较的相关文献报道。中药材商品规格等级的划分是衡量药材质量优劣的重要标准,药材的规格与其指标成分含量存在密切关联[18‑19]。2020年版《中国药典》中以蒙花苷含量作为野菊花药材的质量评价指标,但野菊花富含多种活性成分,仅以蒙花苷作为评价野菊花药材质量的指标不够全面。因此,本研究通过建立野菊花药材指纹图谱测定方法,并结合相似度评价、聚类(CA)分析、主成分(PCA)分析和方差分析等化学计量学方法对不同规格野菊花药材的指纹图谱信息进行分析,比较其差异性,为野菊花药材规格等级的准确划分和质量控制提供实验依据。
1 仪器与试药
1.1 仪器
Waters Arc型高效液相色谱仪(美国Waters公司);XP26型百万分之一天平、ME204E型万分之一天平(瑞士梅特勒‑托利多公司);Milli‑Q Direct型超纯水系统(德国Merck公司);111B型二两装高速中药粉碎机(浙江瑞安市永历制药机械有限公司);TC‑15型套式恒温器(海宁市新华医疗器械厂);KQ‑500DE型数控超声波清洗器(昆山市超声仪器有限公司);Mitutoyo型电子数显游标卡尺(昆山科德三测量仪器有限公司)。
1.2 试药
蒙花苷对照品(批号:111528‑201911,质量分数98.5%)、绿原酸对照品(批号:110753‑202018,质量分数96.1%)购于中国食品药品检定研究院;异绿原酸C对照品(批号:wkq20031101,质量分数99.21%)、新绿原酸对照品(批号:wkq18030107,质量分数98.0%)、隐绿原酸对照品(批号:wkq19120203,质量分数99.5%)、异绿原酸A对照品(批号:wkq20020403,质量分数99.45%)、异绿原酸B对照品(批号:wkq17060705,质量分数98.0%)购于四川省维克奇生物科技有限公司;液相用甲醇、磷酸、乙腈为色谱级(德国默克股份有限公司);水为超纯水,其余试剂均为分析纯。
14批野菊花药材(编号:S1‑S14)经广东一方制药有限公司魏梅主任药师鉴定为菊科植物野菊C.indicumL.的干燥头状花序,产地分别来自安徽、河南和湖北。根据野菊花药材的直径大小,将每批分为3类,以a、b、c分别表示直径0.1~0.3 cm、直径0.3~0.5 cm、直径0.5~1.0 cm,14批野菊花药材根据不同规格共分成42份样品,分别编号为a1~a14、b1~b14、c1~c14。
2 方法与结果
2.1 色谱条件[20]色谱柱:Waters XBridge C18色谱柱(250 mm×4.6 mm,5μm),流动相:乙腈(A)‑0.1%磷酸溶液(B)梯度洗脱(0~16 min,5%~24%A;16~19 min,24%~29%A;19~29 min,29%~30%A;29~34 min,30%~32%A);流速:1.2 mL/min;柱温:30℃;检测波长:326 nm;进样量:10μL。
2.2 溶液配制
2.2.1 对照品溶液的制备 取蒙花苷对照品适量,精密称定,加入甲醇配制成对照品母液A;另取新绿原酸、绿原酸、隐绿原酸、异绿原酸A、异绿原酸B、异绿原酸C对照品适量,精密称定,加入14%乙腈配制成对照品混合母液B;取对照品A、B液混合,置10 mL量瓶中,加入14%乙腈定容至刻度,摇匀,配制成含新绿原酸47.040 0μg/mL、绿原酸89.469 1 μg/mL、隐 绿 原 酸63.406 0μg/mL、异 绿 原 酸A 71.736 0μg/mL、异绿原酸B 65.954 0μg/mL、异绿原酸C 57.722 0μg/mL、蒙花苷56.243 5μg/mL的混合对照品储备液。
2.2.2 供试品溶液的制备 取野菊花药材粉末(过三号筛)约0.3 g,精密称定,置圆底烧瓶中,加水50 mL,加热回流45 min,滤过,滤液蒸干,残渣加30%甲醇100 mL,超声处理(功率200 W,频率50 kHz)30 min,取出,放冷,滤过,取续滤液,即得。
2.3 方法学考察
2.3.1 专属性试验 分别精密吸取空白溶剂、对照品溶液及供试品溶液,照“2.1”项色谱条件进样测定,色谱图见图1。可见,供试品在与对照品溶液相应的保留时间处有相同的色谱峰,且空白溶剂无干扰,表明方法专属性良好。

图1 专属性试验HPLC色谱图Figure 1 HPLCchromatograms of specificity test
2.3.2 精密度试验 精密吸取“2.2.2”项供试品溶液,按“2.1”项色谱条件连续进样6次,以10号峰蒙花苷为参照峰,计算各特征峰的相对保留时间和相对峰面积RSD均小于2.0%,表明仪器精密度良好。2.3.3 重复性试验 取同一批野菊花粉末,精密称定,平行称定6份,按“2.2.2”项方法制备供试品溶液,按“2.1”项色谱条件进样测定。以10号峰蒙花苷为参照峰,计算各特征峰的相对保留时间和相对峰面积RSD均小于2.0%,表明方法重复性良好。
2.3.4 稳定性试验 精密吸取“2.2.2”项供试品溶液,按“2.1”项色谱条件分别在0、2、4、6、8、12、24 h进样测定,以10号峰蒙花苷为参照峰,计算各特征峰的相对保留时间和相对峰面积RSD均小于2.0%,表明供试品溶液在24 h内稳定性良好。
2.4 不同规格野菊花药材的指纹图谱
2.4.1 不同规格野菊花药材指纹图谱的建立 分别取14批不同直径野菊花药材(样品a1~a14、b1~b14、c1~c14),按“2.2.2”项方法制备供试品溶液,按“2.1”项色谱条件进样测定,记录色谱图。不同直径野菊花的样品数据依次采用“中药色谱指纹图谱相似度评价系统”(2012)进行评价,分别生成4个直径HPLC对照图谱,见图2。确定了10个共有特征峰,通过对照品指认,确定了1号峰为新绿原酸,2号峰为绿原酸,3号峰为隐绿原酸,6号峰为异绿原酸B,7号峰为异绿原酸A,8号峰为异绿原酸C,10号峰为蒙花苷。

图2 不同规格野菊花药材共有对照指纹图谱Figure 2 Common control fingerprints of different specifica‑tions of C.indicum
2.4.2 不同规格野菊花药材相似度评价 计算样品a1~a14、b1~b14、c1~c14生成的对照指纹图谱与未划分规格野菊花药材生成的对照指纹图谱的相似度,结果见表1。可见,各批次药材的相似度均>0.90,表明相同直径范围的不同批次野菊花具有较高的相似度。

表1 不同规格野菊花药材指纹图谱的相似度Table 1 The similarity of fingerprints of different specifications of C.indicum
2.4.3 不同规格野菊花药材特征峰比较 以野菊花药材规格为横坐标,该规格下所有样品峰1‑峰10的“峰面积/取样量”均值为纵坐标,对指纹图谱中的10个特征峰随规格的变化趋势进行分析,其中a为直径0.1~0.3 cm,b为直径0.3~0.5 cm,c为直径0.5~1.0 cm,d为未划分规格的野菊花药材。变化规律见图3。可见,各特征峰的“峰面积/取样量”随野菊花直径的增加呈递减趋势,且未划分规格的药材d的规格可能大多集中在0.3~0.5 cm。
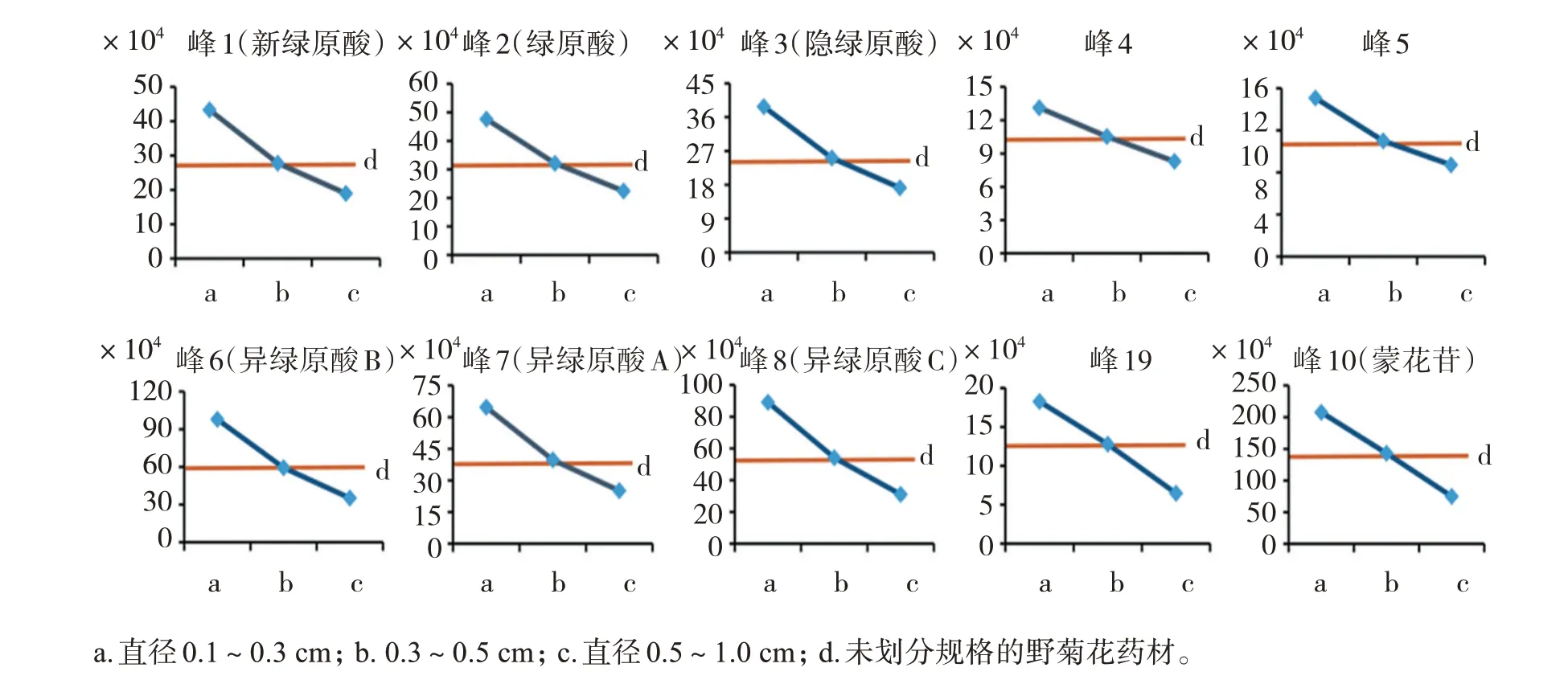
图3 不同规格野菊花药材特征峰比较Figure 3 Comparison of characteristic peaks of different specifications of C.indicum
2.4.4 聚类分析 聚类分析是对样品或指标成分进行分类的一种多变量统计技术,也是应用范围最为广泛的多元统计方法之一[21]。采用SPSS 20.0软件对各特征峰的“峰面积/取样量”进行聚类分析,建立14批野菊花不同直径大小的聚类分析树状图,结果见图4。可见,不同规格野菊花共42个样本聚为2类,由于不同批次间样品存在一定差异,除去个别样品(a4和a6),b与c聚为I类,a聚为Ⅱ类。综上所述,根据14批不同规格野菊花样品指纹图谱聚类分析结果,基本可将直径0.1~0.3 cm(a)与直径0.3~1.0 cm(b和c)两种规格的野菊花区分开。

图4 42批野菊花样品的聚类分析(CA)树状图Figure 4 Cluster analysis(CA)dendrogram of 42 batches of C.indicum
2.4.5 主成分分析 主成分分析是一种广泛应用于多学科的多元统计方法,可用于简化数据,快速实现对数据的可视化识别[22]。采用SPSS 20.0软件对10个共有峰的“峰面积/取样量”进行因子分析,根据主成分的提取原则,取特征值大于1对应的成分作为提取主成分[23],结果见表2、表3。可见,前2个主成分累积贡献率为96.262%,表明提取的2个主成分能基本反映全部指标的信息。主成分1的特征值为9.129,方差贡献率为82.989%,载荷较高的峰有新绿原酸、绿原酸、隐绿原酸、异绿原酸A、异绿原酸B、异绿原酸C,表明这6个峰主要反映主成分1的信息;主成分2的特征值为1.460,方差贡献率为13.273%,载荷较高的峰有峰4、峰5、峰9及蒙花苷,表明这4个峰主要反映主成分2的信息。上述对野菊花药材指纹图谱差异贡献较大的成分,可能是影响野菊花药材质量的关键成分。

表2 主成分分析Table 2 Principal component analysis

表3 主成分因子载荷矩阵Table 3 Loading matrix of principal component factor
2.4.6 方差分析 采用SPSS 20.0软件对42份野菊花样本各特征峰的“峰面积/取样量”进行方差分析,并采用字母标记法进行显著性分析,结果见表4。可见,特征峰1(新绿原酸)、峰2(绿原酸)、峰3(隐绿原酸)、峰6(异绿原酸B)、峰7(异绿原酸A)、峰8(异绿原酸C)、峰9、峰10(蒙花苷)在0.1~0.3 cm、0.3~0.5 cm、0.5~1.0 cm直径范围间差异均有统计学意义;特征峰4在0.1~0.3 cm与0.5~1.0 cm直径范围间差异有统计学意义;特征峰5在0.1~0.3 cm与0.3~1.0 cm直径范围间差异有统计学意义,表明不同直径的野菊花在成分含量间存在较大差异。

表4 不同规格野菊花药材方差分析比较结果(峰面积/取样量,mAu·min−1·g−1)Table 4 Comparison results of variance analysis of different specifications of C.indicum
3 讨论
野菊花中的黄酮类和酚酸类物质均具有清热解毒的药理活性,因此仅以蒙花苷的含量为指标不足以准确、全面地评价野菊花药材质量。中药指纹图谱在一定程度上能基本反映中药材全貌,实现对中药材内在质量的综合评价[24]。目前,采用HPLC建立野菊花药材的指纹图谱分析方法研究较少,本文探讨了野菊花外观性状中直径大小与内在成分之间的关系,通过建立指纹图谱,确定了野菊花药材的10个共有峰,通过与对照品色谱峰比对,能够确定7个共有峰的化学成分。
从本文的指纹图谱比较结果可以看出,各特征峰的峰面积/取样量比值与野菊花药材直径成反比,表明野菊花药材的直径越小,各特征成分的含量越高。2020年版《中国药典》中规定野菊花药材的直径范围为0.3~1.0 cm,实际在市场流通的货源中,不乏直径小于0.3 cm者,这部分药材质量理应更上一层,但混杂在大直径规格的药材中,不利于其临床疗效的发挥。因此针对野菊花药材规格的划分,可以考虑增加小于0.3 cm的规格。当然,直径越小意味着越早采摘,产量可能也会相对较小,但作为商品,如能进行针对性的市场开发,仍应具有较好的市场前景。
聚类分析可将野菊花药材大致分为2种规格:直径0.1~0.3 cm与直径0.3~1.0 cm,并且特征峰的方差分析结果均具有统计学意义;主成分分析结果表明,酚酸类成分集中在第一主成分上,且占有较高的载荷,表明酚酸类成分可代表所匹配大部分的色谱峰。因此,HPLC指纹图谱结合化学计量学可有效评价不同规格野菊花药材质量,反映各成分间的含量差异,对野菊花药材的规格等级的划分、质量评价和临床使用具有一定的参考意义。
