金属掺杂改善HfO2阻变存储器(RRAM)氧空位导电细丝性能
2021-07-28王菲菲代月花卢文娟鲁世斌汪海波万丽娟蒋先伟
王菲菲,代月花,卢文娟,鲁世斌,汪海波,万丽娟,蒋先伟
(1.安徽大学电子信息工程学院,安徽 合肥 230601; 2.电子信息系统仿真设计安徽省重点实验室, 合肥师范学院,安徽 合肥 230601; 3.光电探测科学与技术安徽高校联合重点实验室,合肥师范学院,安徽 合肥 230601)
1 前 言
基于二元金属氧化物的阻变存储器(RRAM)与互补金属氧化物半导体(CMOS)工艺具有良好的兼容性[1]、器件结构简单、高密度、低功耗、编程/擦除速度快等突出优点,成为时下微电子技术领域热点研究对象[1-3]。RRAM利用阻变层在外加电场下发生高、低阻态的可逆转变效应以实现信息存储[4-6],虽然RRAM已经过十多年的快速发展,但由于阻变开关可逆变化的均一性、可靠性不高,尚未获得大规模的产业应用。导电细丝阻变理论是时下研究者普遍承认的阻变机理,通过导电细丝(成分为氧空位或金属离子)的形成与断裂,实现RRAM高、低阻态转变[7-10]。KALANTARIAN等[11]认为导电细丝形状、半径都会影响RRAM的Set-Reset电压、数据保持特性和耐擦写性。因此,理解阻变过程中可逆转变的物理机理是优化器件性能、促进技术发展的有效途径。KWON等[12]通过高分辨率透射电子显微镜发现在 Pt/TiO2/Pt器件中存在氧空位纳米导电细丝;沉淀工艺制作ZnO薄膜层时在(0001)面上产生氧空位,在高电场的作用下,过渡金属氧化物体内产生的氧空位团簇在两个电极之间产生导电细丝,即低阻态现象[13]。近年来,很多利用掺杂手段改善氧空位导电细丝RRAM的性能,从而降低forming/set电压,提高器件的耐擦写性[14-17]。Ni/HfO2/Pt 器件,在低阻态时导电细丝既含有Ni离子也有氧空位(VO)[18],TRAPATSELI等[19]用电压脉冲扫描发现基于TiOX的RRAM,掺入杂质 Al后阈值电压降低,并且在电压低至0.9 V时支持模拟二元开关,研究结果展示了实现低功耗RRAM系统的潜在途径。
第一性原理能够从物理底层获取缺陷的阻变行为来解释实验现象,MAGYARI-KöPE等[20]使用第一性原理研究了当NiO、TiO2晶体内有氧空位缺陷时,禁带中出现大量杂质能级,该缺陷可以辅助电子传输,增强导电能力,导致低阻态;当外加电场时,氧离子移动到有序的氧空位中导致氧空位混乱排列即所谓的高阻态。文献[21]计算λ-Ta2O5中氧空位的迁移势垒和形成能,将微观理论与宏观实验相结合,用缺陷的迁移势垒解释了器件实际操作速度快慢的原因。虽然基于密度泛函理论对HfOx薄膜的电子结构进行了广泛研究[22-23],但氧空位在阻变材料体内如何团簇;导电细丝在阻变材料体内如何选择生长路径;当外加电场时,氧空位如何重新排列等都是值得进行深入研究的问题。
本研究使用第一性原理定量分析研究氧空位在HfO2体内形成导电细丝的过程,并且分析了缺陷对材料电子结构的影响;基于导电细丝模型,系统研究了掺入不同金属杂质(Ag、Mg、Ni、Cu、Al、Ta、Ti)对缺陷体系电子结构的影响。
2 计算模型与方法
单斜HfO2(m-HfO2)在常温下比四方、立方晶向稳定,被认为是最有潜力的阻变材料。本研究采用Materials Studio软件建立m-HfO2结构:空间群号P21/c,键长分别为:a=5.117 Å,b=5.1754 Å,c=5.2915 Å的12个原子的单胞[24],模型如图1(a)所示,与实验数据a=5.116 Å,b=5.172 Å和c=5.295 Å 基本相同[25]。将单胞沿XYZ方向扩展成2×2×2共96个原子的超胞体系进行计算,结构如图1(b)所示。
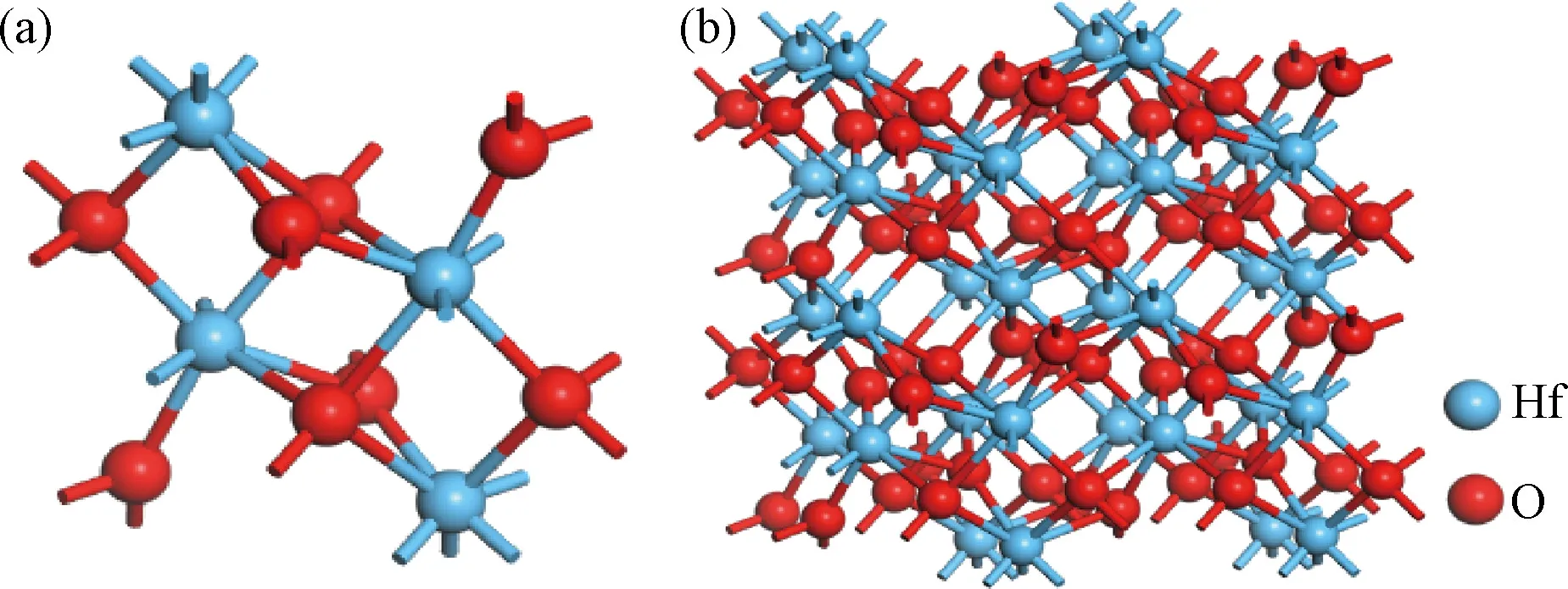
图1 结构模型 (a) m-HfO2单胞; (b) 96个原子的超胞Fig.1 Schematic configuration (a) m-HfO2; (b) super cell
根据计算目的不同,在上述超胞的基础上建立不同的模型,Vo缺陷由去除超胞中的氧原子获得。本研究运用基于密度泛函理论(DFT)的Vienna ab-initio simulation package(VASP)进行结构豫驰及能量、分波电荷密度(PCD)及态密度(DOS)等计算[26]。计算时,电子间的交换关联能通过广义梯度近似(GGA)的PBE方案,经过测试,布里渊区对应的K点设置为3×3×3网格,平面波截断能设置为500 eV。在结构豫驰的过程中,不改变元胞体积,原子间相互作用力的收敛精度为0.015 eV/Å,能量收敛精度为10e-5 eV/atom。
3 结果分析与讨论
3.1 HfO2中氧空位导电细丝的形成
氧空位在外加电压驱动下形成导电细丝的难易程度与氧空位的形成能有密切关系,形成能越低,导电细丝越容易形成,器件形成电压(FV)越低。文献[27]提及氧空位在晶界上的形成能比晶体HfO2体内形成能小,为了找出氧空位在HfO2超胞中最稳定的结构,考虑周期性因素,在(101)面表层选取四个氧空位,位置如图2(a)所示,用VO1~VO4表示,图2(b)为模型俯视图,其中VO1,VO2为三配位氧空位,VO3,VO4为四配位。根据式(1)计算氧空位形成能,其中E(nVoHfO2)表示含有缺陷的超胞体系总能量,E(HfO2)为纯净m-HfO2的总能量,μ0表示氧原子化学势,本研究计算时取值为氧分子能量的一半,n为氧空位的个数[28],计算结果见表1。其中 VO4形成能最低,最容易形成氧空位,与先前计算的四配位氧空位的形成能相差不大[27]。以 VO4作为第一个氧空位,在此方向上沿着下一个可能氧空位的位置,如图2(c)中绿色原子所示,再一次计算团簇形成能[13],发现VO23号氧空位形成能最低,将VO4-VO23看作一个整体,同样方法,VO4-VO23-VO34氧空位团簇在列表中的形成能最低,第四个为 VO46空位,计算结果表明在 [010]晶向上形成近似一排氧空位导电细丝,如图2(f)所示。
Eint=E(nVOHfO2)-E(HfO2)+nμO
(1)
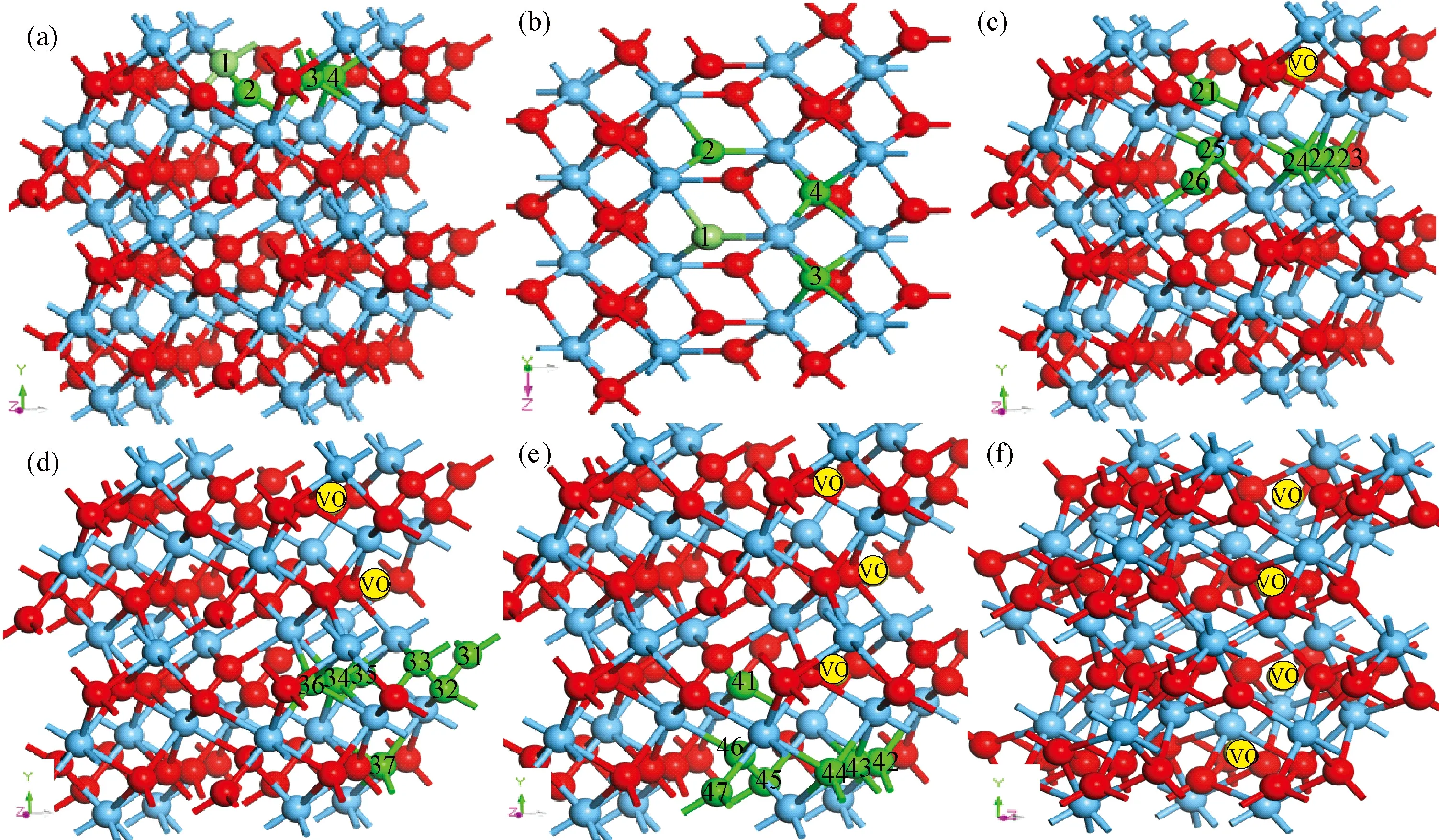
图2 (a) 第一个可能形成的氧空位VO1-VO4; (b) 俯视图; (c) 第二个可能的氧空位VO21-VO26; (d) 第三个可能的氧空位VO31-VO37; (e) 第四个可能的氧空位VO41-VO47; (f) [010]晶向上形成的一排氧空位簇(其中:绿色球表示可能形成氧空位的氧原子,黄色球表示氧空位)Fig.2 (a) The initial VO at one of the positions VO1-VO4; (b) the top view; (c) the second VO might exist position VO21-VO26; (d) the third VO might exist position VO31-VO37, (e) the fourth VO might exist position VO41-VO47; (f) a row VOS of [010] crystal orientation(Green balls represent possible VO,yellow ball indicates VO)
纯净的HfO2是宽禁带材料,几乎不导电,但氧空位的存在改变了体系的电子结构,根据缺陷在禁带中产生的杂质能级,当最大等势面值取0.004时,分别计算Hf32O63(一个氧空位VO4)、Hf32O62(两个氧空位VO4-VO23)、Hf32O61(三个氧空位VO4-VO23-VO34)及Hf32O60(四个氧空位VO4-VO23-VO34-VO46)四种缺陷体系的PCD,如图3所示。从图中可以清晰的看出在氧空位周围聚集着黄色电荷,但仅在 Hf32O60(四个氧空位 VO4-VO23-VO34-VO46)缺陷体系中黄色电荷将上下两个面连在一起形成导电通道,体系呈低阻态,从而改变材料的导电性。对含有四个氧空位缺陷体系的电荷DOS进行计算,结果如图4所示。与纯净的 HfO2相比,费米能级相对与价带顶来说更靠近导带底,禁带中出现三条杂质能级带,且在费米能级处峰值最高,表示材料带隙的能量降低,这些杂质能级的产生使得价带中的电子很容易的向导带跃迁,从而改变材料的导电性。从计算结果发现价带的DOS主要由体系中的O贡献、导带则主要由体系中的Hf贡献,同时禁带中杂质能级基本由Hf原子贡献。杂质能级的存在导致材料发生阻变行为,相对于纯净的HfO2材料体系,其电学特性得到明显改善。
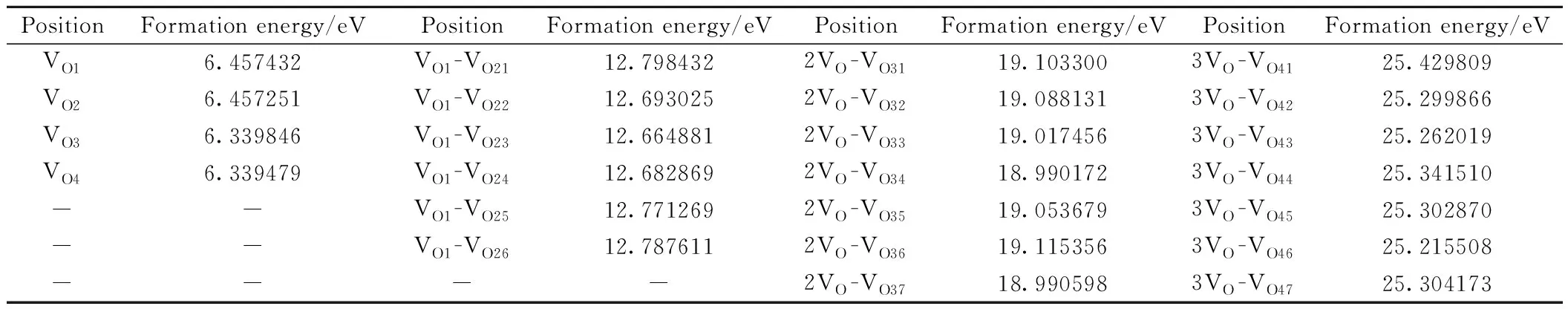
表1 不同位置氧空位的形成能Table 1 Formation energy of oxygen vacancy in different positions

图3 四种不同VO浓度的分波电荷密度图(a)~(d)分别代表Hf32O63、Hf32O62、Hf32O61及Hf32O60Fig.3 Partial wave charge density of defective system with different VO concentration (a)-(d) represent Hf32O63, Hf32O62, Hf32O61 and Hf32O60, respectively
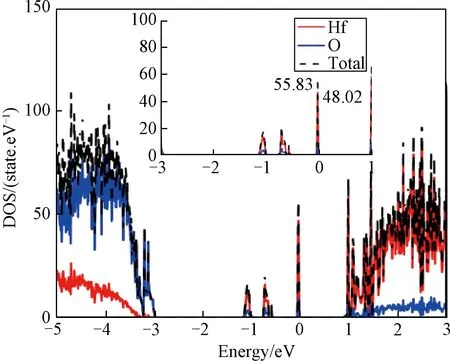
图4 含4个氧空位缺陷体系的态密度Fig.4 Total electron Density of states (DOS) of the bulk HfO2with four oxygen vacancies(4Vo)
虽然仍未清晰的理解RRAM阻变机理,但大家普遍接受阻变特性与导电细丝有着密切的关系。在缺陷体系中杂质能级产生的原因,值得对缺陷体系进一步深入分析。对32个Hf原子态密度计算,如图5(a)所示,其中黑色能级为体系中32个Hf原子产生的态密度,红色为67/68/73/74/75/76/78/81/82/89/90/号Hf原子产生,可以发现红色和黑色基本重合,进一步分析发现上述Hf原子都位于氧空位周围,位置如图5(b)所示,而远离氧空位的Hf不产生杂质能级如图5(a)中插图所示。图5(c)为氧空位周围Hf原子的分波态密度,结果表明对杂质能级的贡献主要是由氧空位周围Hf原子的d轨道,可能由于氧原子具有很强吸附电子能力将Hf最外层轨道上电子局域,氧空位形成后,不受氧原子的束缚,从而Hf 的d轨道杂化产生杂质能级,上述结果表明氧空位的存在使周围Hf原子产生悬挂键,能显著改变材料的电学特性。
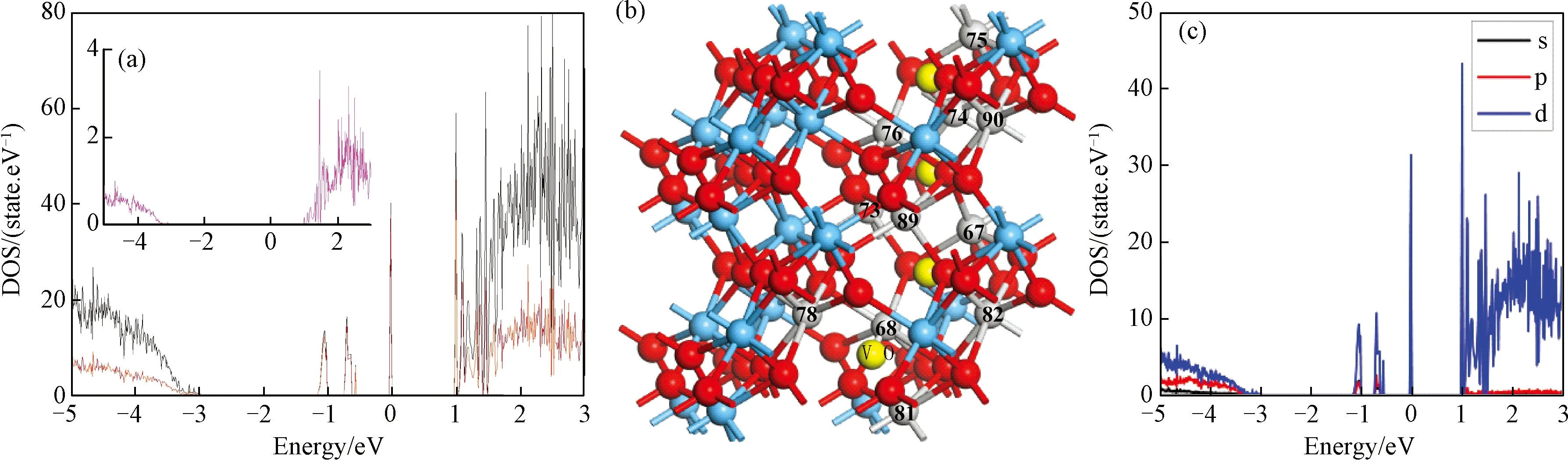
图5 (a)缺陷体系中Hf原子态密度,插图为远离Vo的Hf原子态密度;图(b)Vo周围Hf原子位置,灰色球所示;(c)VO附近Hf原子分波态密度(PDOS),图中黑色表示s轨道,红的表示p轨道,蓝色表示d轨道Fig.5 (a) DOS of Hf in the defect HfO2, The illustration is produce by Hf away from Vo; (b) gray balls represent of the Hf near VO; (c) the projected electron density of states (PDOS) of Hf near VO, black lines represents the s orbital, red lines represents the p orbital, the blue lines represents the d orbital
3.2 不同金属杂质对氧空位导电细丝的影响
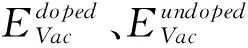
(2)
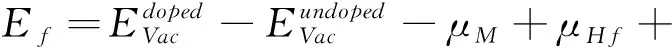
(3)
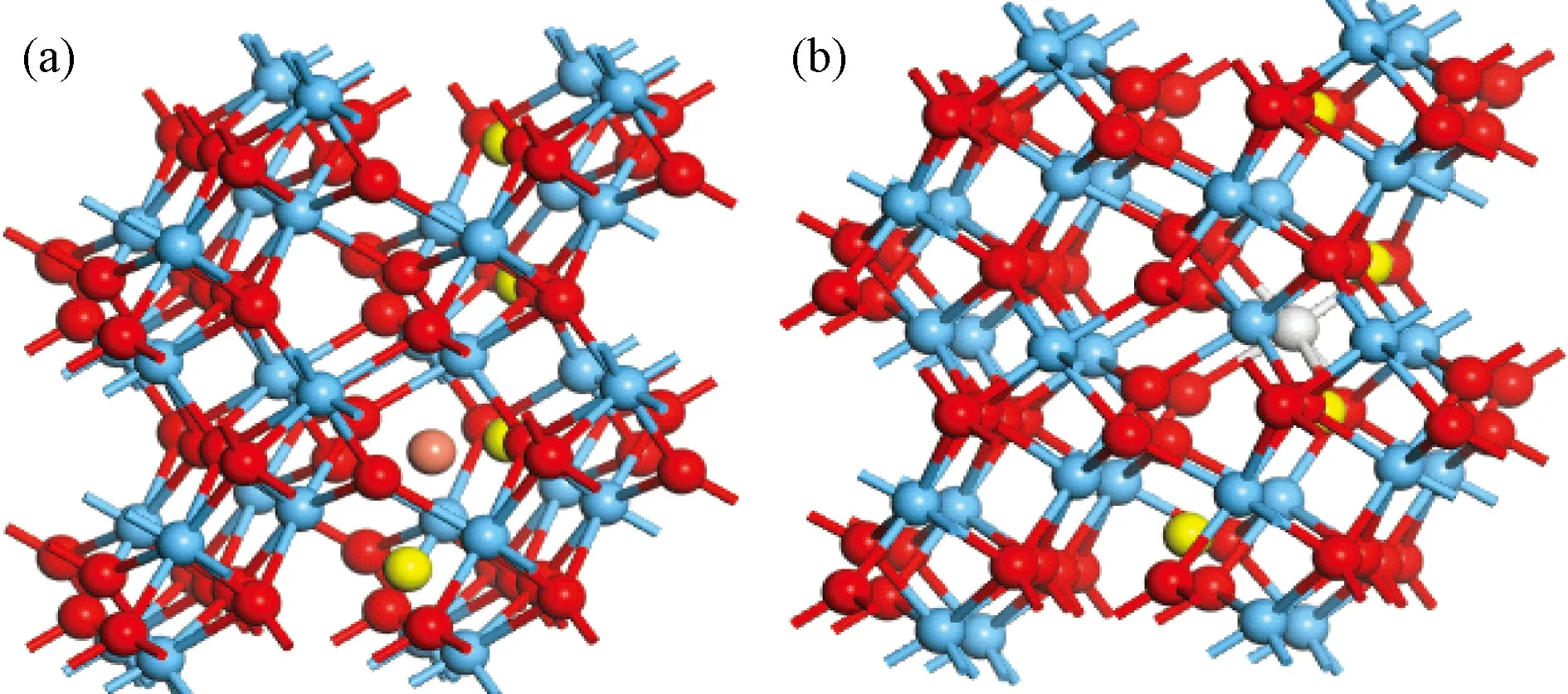
图6 结构模型,橙色代表间隙金属,灰色代表替位金属(a) 间隙式; (b) 替位式Fig.6 Orange ball represent the interstitial metal, the gray represent substitute metal (a) interstitial doping; (b) substitute doping

图7 7种常见金属在缺陷体内形成能Fig.7 Seven common metals in a defective bulk
3.2.2共掺杂体系态密度及PCD分析 金属Ni形成能最低说明Ni在体内最容易存在。最高等势面的值可以反映导电细丝形的难易程度[25],值越大,说明导电细丝越容易形成,通过计算得出掺入不同金属后形成导电细丝的最高等势面值,如图8所示。图9为在最高等势面下,导电细丝的形状,通过比较可以看出掺入金属Ni后,缺陷体系导电细丝的最高等势面值最大。文献[30]通过实验研究分析掺入不同金属可以改善器件的FV和ON/OFF ratio,文中说明不同金属对器件的FV有不同的影响,掺入金属Ni使器件的形成电压最小,而W的掺入却相反,增加开启电压值。FV形成电压越低,器件的功耗越低,与本研究的计算一致。对VO-Ni共缺陷体系态密度进行分析,结果如图10所示,与没有掺入金属杂质(图5)相比,共掺杂体系能带右移,在费米能级处,杂质能级的量子态密度基本相同,并且可以看出是由氧空位缺陷所产生。但Ni的掺入,导致价带顶和费米能级之间多了若干条杂质能级,从计算的结果发现是由Ni贡献,且在-0.85 eV附近峰值很尖锐,并且Ni原子的引入使氧2P态电子产生的价带顶右移0.5 eV,Hf 3d态产生的导带底右移0.5 eV,这些改善了半导体材料的电学特性。根据公式Eint=E2+E0-2E1,其中Eint表示缺陷间的相互作用能,E0表示完整超胞总能量,E2为含有两种缺陷时能量,E1一个缺陷时超胞能量[13],计算出Ni与氧空位导电细丝相互作用能为-2.335 eV,说明缺陷之间相互吸引,而这种相互吸引作用影响了缺陷体内的电子特性,Ni金属对氧空位导电细丝周围的价电子具有较强的局域作用,从而在周围形成较为均匀的导电细丝,这些改变有利于优化阻变存储器的形成电压和器件的均匀性。通过分析PDOS可以发现在禁带中产生的杂质能级,不再仅是氧空位周围Hf原子的,同时Ni的d轨道电子同样在禁带中产生杂质能级。当Ni原子离的较远时,模型如图11(a)所示,因为晶胞体积的原因,金属对此体系仍然有一定的影响,当最高等势面最大值为0.006时,PCD如图11(b)所示,比较发现最高等势面最大值比不掺杂时仅大0.0015。可以推断出当金属与氧空位距离越远,对导电细丝的影响将越来越弱。
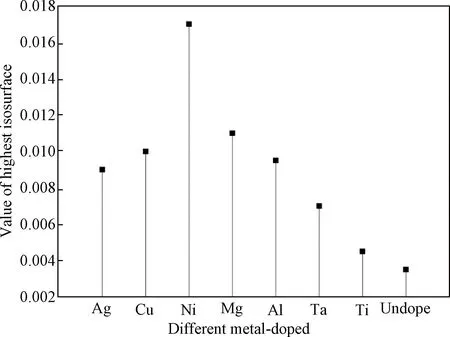
图8 不同金属掺杂共缺陷体系的最高等势面值Fig.8 Value of highest isosurface of different metal Doping systems
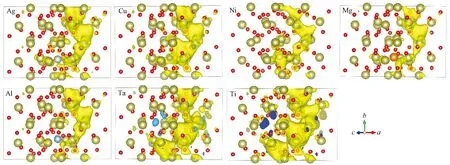
图9 不同金属的共缺陷体系在最高等势面值下电荷分波密度图Fig.9 Partial wave charge density of seven different Co-doped models at highest isosurface
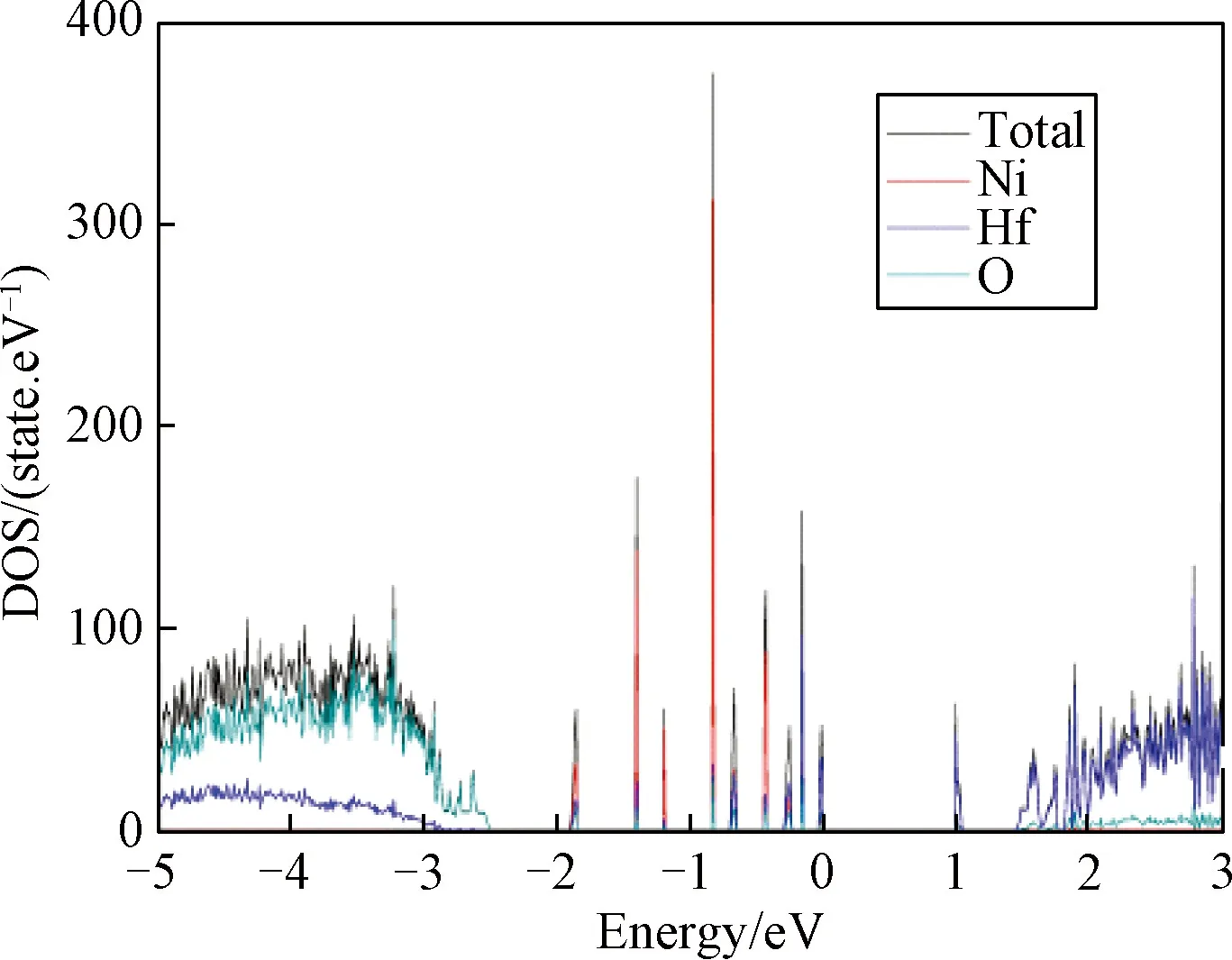
图10 共缺陷体系态密度Fig.10 DOS of Co-doped defect system
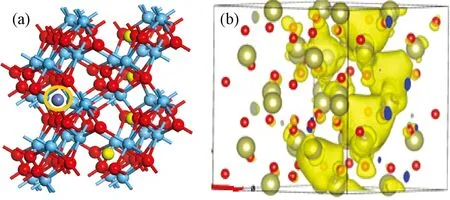
图11 (a) Ni原子较远模型,蓝灰色为Ni原子; (b)共掺杂体系分波电荷密度Fig.11 (a) Model of Ni far away from CF; (b) Partial wave charge density of Co-doped defect system
本研究基于密度泛函理论的第一性原理模拟计算软件,从微观机理上计算分析m-HfO2阻变材料中的氧空位导电细丝的形成过程,及共掺杂情况下金属对缺陷体系电子结构的影响。
1.计算模拟出HfO2超胞体系在[010]晶向上4个氧空位排列形成导电细丝,DOS计算结果表明禁带中产生一定数量的杂质能级是由氧空位周围67/68/73/74/75/76/78/81/82/89/90/号Hf原子产生,而体内其它Hf原子对杂质能级基本没有影响,PDOS计算结果表明,杂质能级由Hf的d轨道产生。这意味着氧空位缺陷对周围Hf的电子结构产生较大影响,从而被称之为氧空位导电细丝。
2.对氧空位导电细丝体系掺入7种金属原子,研究发现Ni原子在缺陷体内最容易存在,在7种共掺杂体系中最高等势面值最大,体系的分波电荷密度更均匀。Ni与氧空位两种缺陷间的相互作用能为-2.335 eV,相互吸引,从电荷密度图中可以发现Ni的引入在价带顶和费米能级之间产生若干条杂质能级,在-0.85 eV峰值最尖锐,改变了复合缺陷体的电子性能。
3.当Ni 距离较远时,对氧空位导电细丝体系性能的改善效果不明显。
