Growing and aging of hematopoietic stem cells
2021-07-24IonUdroiuAntonellaSgura
Ion Udroiu, Antonella Sgura
Ion Udroiu, Antonella Sgura, Department of Science, Roma Tre University, Rome 00146, Italy
Abstract In the hematopoietic system, a small number of stem cells produce a progeny of several distinct lineages.During ontogeny, they arise in the aorta-gonadmesonephros region of the embryo and the placenta, afterwards colonise the liver and finally the bone marrow.After this fetal phase of rapid expansion, the number of hematopoietic stem cells continues to grow, in order to sustain the increasing blood volume of the developing newborn, and eventually reaches a steady-state.The kinetics of this growth are mirrored by the rates of telomere shortening in leukocytes.During adulthood, hematopoietic stem cells undergo a very small number of cell divisions.Nonetheless, they are subjected to aging, eventually reducing their potential to produce differentiated progeny.The causal relationships between telomere shortening, DNA damage, epigenetic changes, and aging have still to be elucidated.
Key Words: Bone marrow; Fetus; Growth and development; Hematopoietic stem cells; Leukemia; Liver
INTRODUCTION
Different kind of blood cells (erythrocytes, platelets, macrophages, neutrophils, eosinophils, basophils, dendritic cells, B-cells, T-cells, and NK-cells) are continuously produced throughout life.Being responsible for oxygen and carbon dioxide transport, immune response, and haemostasis, blood cell production is regulated on demand upon metabolic needs, pathogen invasion, and injuries.This process of differentiation and maturation is called hematopoiesis: A small number of cells, called hematopoietic stem cells (HSC), generate several distinct lineages (through intermediate progenitors and precursors), while maintaining a precise balance between self-renewing and differentiative divisions.In humans, this hierarchically-organized system generates two million red blood cells every second[1].
Several works on the ontogeny of the hematopoietic system have used zebrafish, amphibian, and avian models[2], but studies on the development of murine hematopoiesis have given most of the decisive insights in relation with human ontogeny[3].This is even more true for studies dealing with aging of HSC, which used almost exclusively murine models[4].
In this review, we will deal with the growth of the HSC pool (not only during the fetal stage, but also in infancy) and the dynamics during aging, combining information from human and murine studies.
PRENATAL GROWTH OF THE HSC POOL
Hematopoiesis first rises in the blood islands of the yolk sac, producing mainly erythrocytes, but also megakaryocytes and macrophages[5].This process is termed “primitive erythropoiesis”, as it produces large, nucleated erythrocytes that express embryonic hemoglobins (called primitive erythrocytes).This first hematopoietic wave does not comprise HSC, as defined by the potential to reconstitute hematopoiesis in an adult[6].Therefore, while definitive hematopoiesis is a permanent process deriving from self-renewing HSC, primitive hematopoiesis takes place just once in the yolk sac.The precursors producing primitive erythrocytes appear in mouse at embryonic day (ED) 7.25 and disappear after ED 9[3].This period corresponds, in human, to gestation weeks (GW) 2.5-4.
In the same yolk sac, a second hematopoietic wave begins at the start of somitogenesis (mouse ED 8.25), comprising precursors of the myeloid and erythroid lineages.Erythrocytes produced by this wave are anucleated (like in the adult) and therefore called “definitive erythrocytes”.Nonetheless, also this hematopoietic wave of erythromyeloid progenitors does not comprise HSC[5].
The third wave of hematopoiesis arises in the placenta (mouse ED 9) and, soon after, in the aorta-gonad-mesonephros (AGM) region, the first intra-embryonic site of definitive hematopoiesis.In fact, the hemogenic endothelium of the dorsal aorta generates the first HSC, with the ability to reconstitute hematopoiesis in an adult, and with B and T lymphoid, myeloid, and erythroid potential[6].This period of HSC expansion in the murine placenta and AGM (mouse ED 9-13) corresponds to GW 4-5 in humans.Indeed, Tavian and Péault[7] observed hematopoietic cells in the AGM of human embryos from gestational days 27 to 42.On the other hand, Muenchet al[8] found hematopoietic stem/progenitor cells in human chorion from 5thto 42ndGW, but only those from 15-24 GW were capable of multilineage and long-term reconstitution when transplanted in immunodeficient mice.
The liver diverticulum emerges at ED 9 in mouse (24thday in humans) and is immediately colonised by yolk sac-derived precursors and later by AGM derived HSC[5].By murine ED 13.5, the fetal liver becomes the major hematopoietic organ; at this stage, the embryonic phase of hematopoietic ontogeny is considered finished.The rich microenvironment represented by the hepatic niche sustains a considerable expansion of HSC between ED 12.5 and 16.5.Indeed, the establishment of hepatic erythropoiesis is witnessed by the appearance of reticulocytes (immature enucleated erythrocytes) in the fetal circulation[9].Beginning from ED 14.5, the absolute number of hepatic HSC starts to decrease, as they migrate (through the circulation) to seed the spleen and later (ED 18.5) the bone marrow[5].In humans, colonization of bone marrow by liver HSC probably begins after GW 9, while opposite opinions exist in relation to the existence of a splenic phase[10-12].
POST-NATAL EXPANSION OF THE HSC POOL
The overwhelming majority of the works on the ontogeny of hematopoiesis studied the embryonic and fetal periods.The post-natal development of the hematopoietic system has received much less attention, with the differences between infant and adult hematopoiesis considered only quantitative (differential white blood cells count, number of red blood cells, and percentages of reticulocytes).Nonetheless, these differences underlie specific characteristics of HSC, which make the infant phase distinct from the adult one.
Reticulocytes are immature erythrocytes that, in normal conditions, have a fixed lifespan (the time to develop into mature erythrocytes,i.e.48 h in mice).Thus, their population has a simple kinetic, being a dynamic equilibrium between erythroblasts becoming reticulocytes and reticulocytes becoming erythrocytes.As the total number of reticulocytes in an animal represents the output of its hematopoietic system, it is assumed to be directly proportional to the number of HSC[13].Thus, knowing the total number of reticulocytes at various pre- and post-natal ages, the growth curve of HSC in mouse can be derived (Figure 1A).In the pre-natal phase, it is in perfect agreement with experimental data of Ema and Nakauchi[14], which found a 38-fold increase between ED 12.5 and 16.5.After the first phase of exponential increase, corresponding to the phase of hepatic hematopoiesis, the growth rate decreases, reaching a plateau 21 d after birth.This seems to indicate that, in mouse, the HSC pool stops to grow 3 wk after birth.Interestingly, this agrees with the findings of Bowieet al[15], which found that during the third week after birth, the HSC pool switches from a predominantly cycling population to a mostly quiescent one.From the growth of HSC (Figure 1A), it can be derived their rate of replication (Figure 1B).The exponential decrease of the rate of HSC replication is the same as the one detected in humans[16], although with a different timing (Figure 1C), and is reflected by the rate of telomere shortening in leukocytes (Figure 1D).
Timing of hematopoietic ontogeny is different between mouse and human, and this can be clearly viewed by the time of switching from liver to bone marrow hematopoiesis (at half gestation in humans, but at birth in mice).However, the general process is clearly the same.This opens interesting perspectives, in particular for the studies on the onset of infant leukaemias.A link between the expansion of HSC during ontogeny and the aetiology of leukaemias has been proposed by some authors[9,17-19].Moreover, in infant mice, beside the increase of the quiescent fraction of HSC, Benzet al[20] demonstrated that the percentage of the lymphoid-deficient subpopulation of HSC rapidly rises before birth, remains stable during the first 3 postnatal weeks, and rises again between the third and the fourth week.An ontogenetic parallel between this development of HSC subsets and blood cell production has been showed: Myeloid-lineage cells are predominant in murine fetal blood, being outpaced after birth (when liver hematopoiesis ceases) by an interleukin-7-independent Blymphopoiesis, which lasts until the third-fourth week after birth, when it is replaced by production of B-lymphocytes in a rigorously interleukin-7-dependent manner[18].Although there are no studies on the HSC subsets in children, we can speculate that the pattern is the same as in mice, since the trajectory of the production of blood cell type seems the same[21].Again, this developmental schedule gives interesting parallels to the types of childhood leukaemias.The incidence rates of acute myeloid leukemia peaks in the first year of life and then declines drastically.Moreover, all fetal and neonatal leukaemias are myeloid[22], indicating that the fetal period is specifically associated with this kind of malignancy.On the other hand, acute lymphoid leukemia peaks between 1 and 4 years and then decline: It is tempting to link this phenomenon with the above-mentioned period of sustained lymphopoiesis, which in humans is mirrored by the fact that lymphocytes are the most numerous leukocyte type during the first 4 years of life.
AGING OF HSC
During ontogeny (both pre- and post-natal), blood mass grows allometrically with body mass.As we have seen in the previous sections, also the HSC pool grows, in order to produce the blood cells required by the growing body.Once adulthood is reached, and growth stops, the HSC pool reaches a steady state.The hierarchical organization of hematopoiesis allows the production of hundreds of billions of blood cells per day from a small number of HSC.In this way, HSC undergo a small number of cell divisions, which are divided between differentiation and self-renewal[1].As said before, most adult HSC are quiescent, and divide very rarely.Nonetheless, with each cell division, HSC undergo aging, eventually reducing their potential to produce differentiated progeny[23].This has been demonstrated not only in experiments with mice[24], but also by the reduced transplantation success of HSC isolated from the bone marrow of older human donors[25].
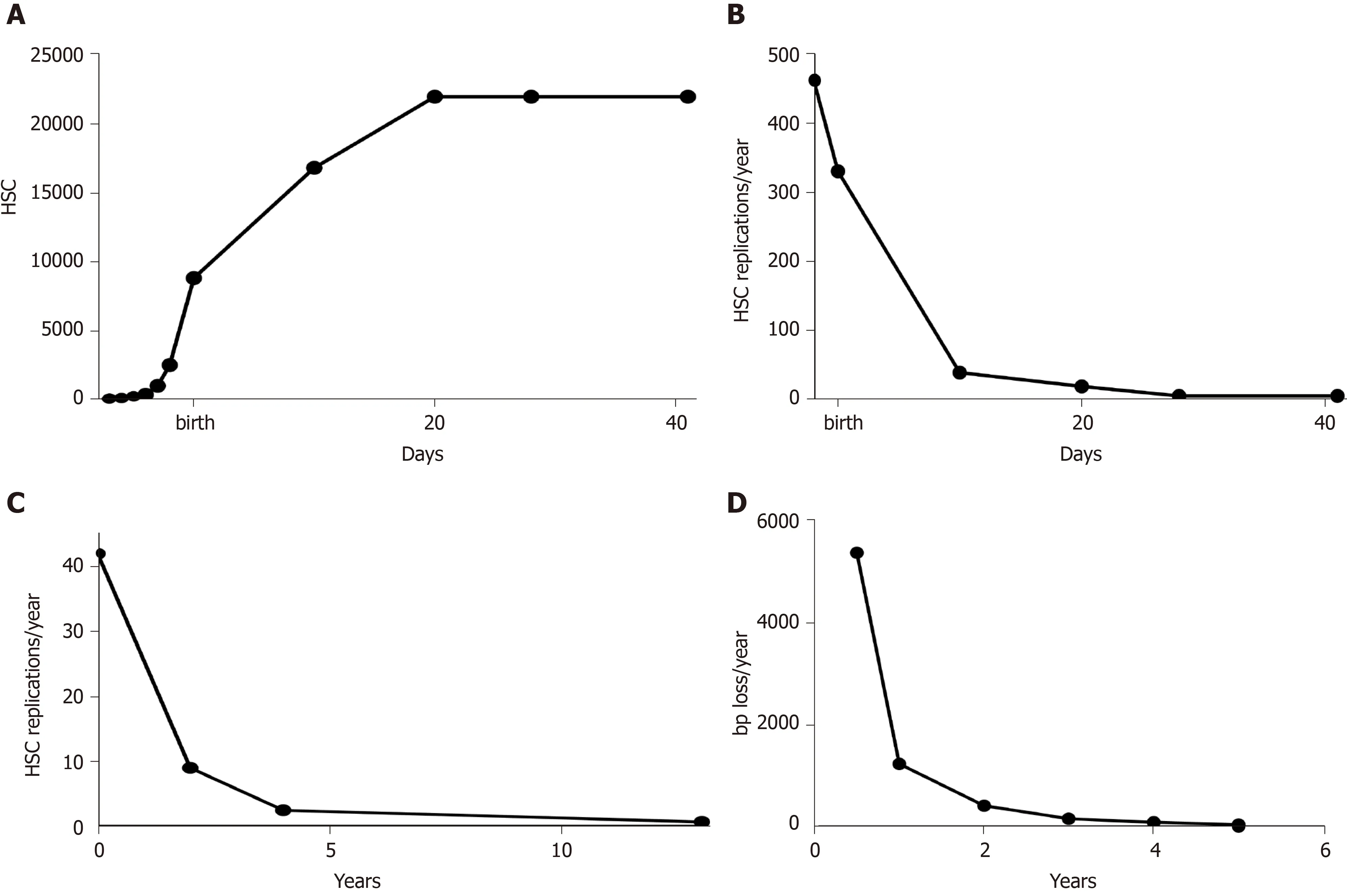
Figure 1 Growth curves in mouse.
We must underline that this “replicative aging” not necessarily corresponds to chronological aging.In other words, HSC age at different paces in different individuals (and species).Indeed, we found a correlation between rate of erythropoiesis, HSC aging, and onset of immunosenescence among mammalian species[26].
The largely quiescent, steady-state, in fact, can be disrupted by an increased demand of blood cells (haemorrhage, infections,etc.), which pushes HSC to produce more progenitors and thus to divide more frequently.
Several phenomena are linked to aging of HSC: DNA damage, telomere erosion, epigenetic reprogramming, impaired autophagy, and mitochondrial activity[23].However, these correlations do not prove a clear link of causality,i.e., we cannot say with certainty which are the primary causes and which by-side effects.
There is general consensus that aging of HSC eventually leads to a reduction of their proliferative potential[23].This can be viewed in the “anaemia of the elderly”,i.e., the well-known reduced production of erythrocytes in people over 60 years of age.Moreover, aging of HSC causes myeloid-skewed hematopoiesis, thus leading to a reduced production of lymphocytes[24].Also this manifestation is well-known in hematology (which has been termed in recent years as immuno-senescence), and has been recognized not only in humans, but also in domestic species such as the horse[27].
Exhaustion of HSC is not a simple, linear process, in which stem cells age, die, and become less and less in number.It may sound counter-intuitive, but as they age, HSC do not diminish, instead they increase (Figure 2).In fact, it has been demonstrated that aged mice display an overall increase in the number of HSC, but these have an impaired functionality[23].The same pattern has been shown in humans[28].Once again, it is interesting to look at the quantity of reticulocytes in the circulation: During aging their number increases (Figure 2), reflecting an accelerated production of red blood cells.Summing up, we can propose this scheme: As HSC age, there is a decline in blood production, which, in turn, stimulates HSC to proliferate; in this way, their number increases, but they age even more (and become even less productive), because they are undergoing more replication; moreover, the increase in number is not accompanied by an equal increase in blood production (because the fraction of inefficient HSC increases), so the stimulus to proliferate continues (Figure 3).
MECHANISMS OF AGING IN HSC
Telomere shortening
It is ascertained that the HSC pool has a finite lifespan.As shown in Figure 1D, HSC replication determines telomere shortening, which in adult humans is estimated to be around 50 base pairs per year[16].In the last 25 years, many authors elected telomere shortening as the mechanism that leads to the end of HSC life[29].This interpretation is based on the well-known Hayflick’s limit, which was discovered in studying the limited lifespan of fibroblasts[30].Thus, at each mitosis undergone by HSC, their telomeres shorten, eventually reaching a critical length and ending their life (by senescence or apoptosis).This simple interpretation, however, can be subjected to many criticisms.First, telomeres shorten in the absence of telomerase, but telomerase is present in all murine cells[31] and in human HSC[32].Therefore, replicative aging due to telomere shortening should be absent in HSC.A counter-argument can be that the activity of telomerase in these cells can limit, but not abolish, telomere shortening.Indeed, there are so much direct and indirect evidence that HSC telomere shortens both in human and mouse, and that progressive reduction of telomere length cannot be negated.Nonetheless, telomere shortening in the presence of telomerase should be linked to another (less investigated) issue: The extent of telomere shortening.During DNA replication, 50-100 bp within each telomere are unable to be copied and are lost[33].However, adult murine HSC losein vivomore than 5000 bp per year[34], which means more than 600 bp lost per replication.This extent of telomere shortening cannot be explained only by the bp that are lost because they are not copied during DNA replication.In fact, von Zglinickiet al[35] demonstrated that in human fibroblasts grown in hyperoxia (40% O2), the rate of telomere shortening is more than 500 bp per replication, compared to 90 bp per replication under 20% oxygen.It has also been demonstrated that hydrogen peroxide-induced oxidative stress determines telomere shortening[36].
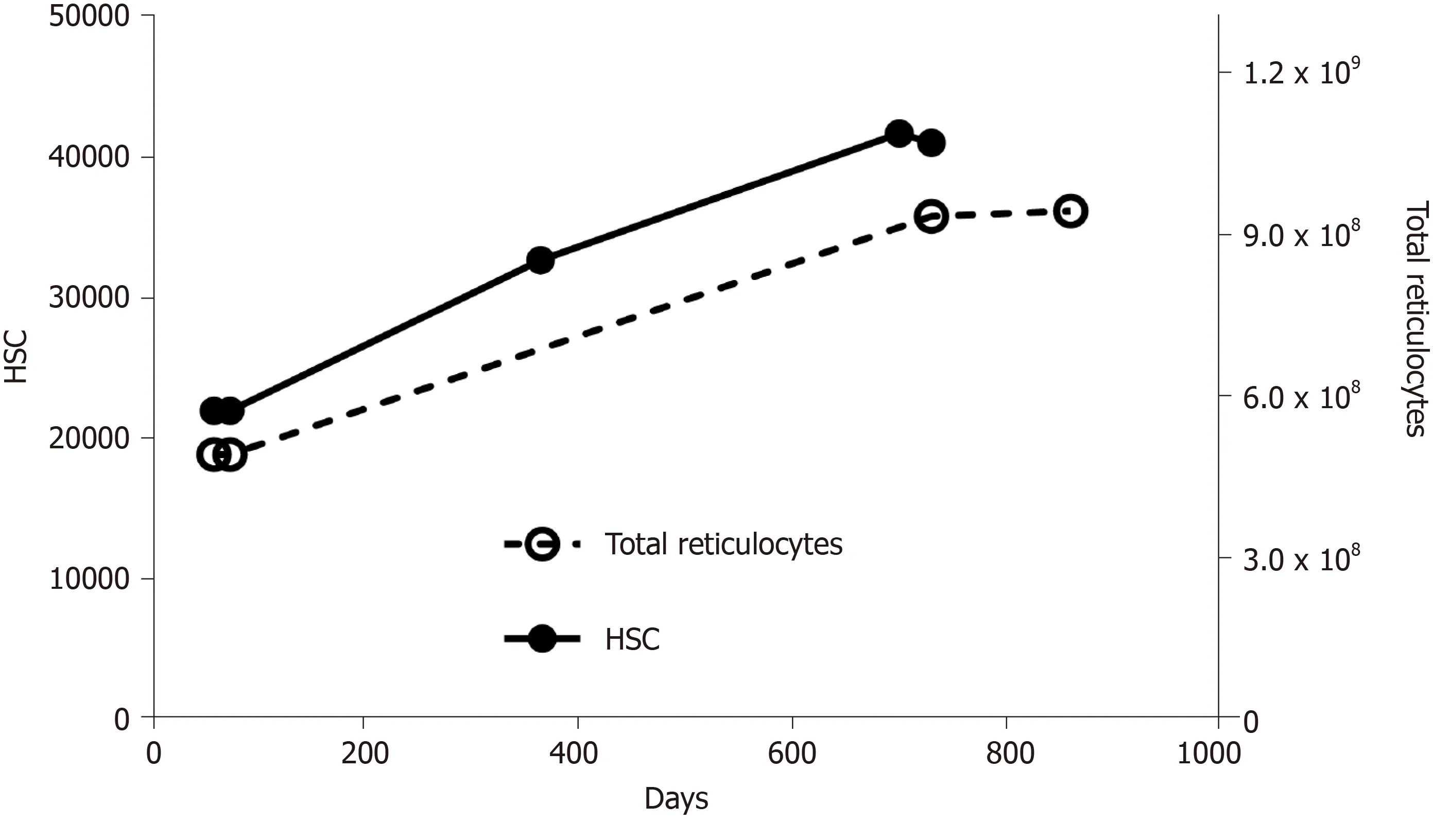
Figure 2 Numbers of hematopoietic stem cells and total reticulocytes in aging mice.
We can infer that HSC, independently of the presence or absence of telomerase, undergo telomere shortening during aging because of oxidative stress.Although the bone marrow (where hematopoiesis takes place) is not a hyperoxic, rather a hypoxic environment, an increase of oxidative stress (through reactive oxygen species production) in the bone marrow hematopoietic niche through aging has been proposed[25].
The second, and more important, objection to the application of the Hayflick’s limit to HSC regards telomere length in mice.It has been demonstrated that cells undergo senescence or apoptosis when telomeres reach a critical length of 1-2 kb[37]: Below this length, telomeric DNA cannot form the characteristic t-loop, remains as an open end without protection of the shelterin complex, and is recognized as a double strand break.However, laboratory mice (which possess very long telomeres) even towards the end of their life still have telomeres longer than 30 kb[34].This seems to indicate that HSC exhaustion is not triggered by short telomeres.
DNA damage
Besides telomere shortening, other mechanisms seem to contribute to the aging phenotype of HSC.One of the most important causes of HSC aging seems the accumulation of random DNA damage.Experimental proof comes from mice deficient in genomic maintenance pathways including nucleotide excision repair (XPD-/-) and nonhomologous end-joining (Ku80-/-)[38].Although deficiencies in these pathways do not deplete HSC numbers with age (which on the contrary increase, in agreement with the model in Figure 3), their functional capacity is severely reduced, leading to loss of reconstitution and proliferative potential, diminished self-renewal, increased apoptosis and, ultimately, functional exhaustion.The same authors demonstrated that endogenous DNA damage increases with age in wild-type HSC[38].As further evidence, accumulated signatures of DNA damage (including γ-H2AX foci) have been detected in HSC of aging mice[39].Moreover, one of the best-known bone marrow failure syndromes is Fanconi anaemia, a genetic disease characterized by defective DNA repairviahomologous recombination.
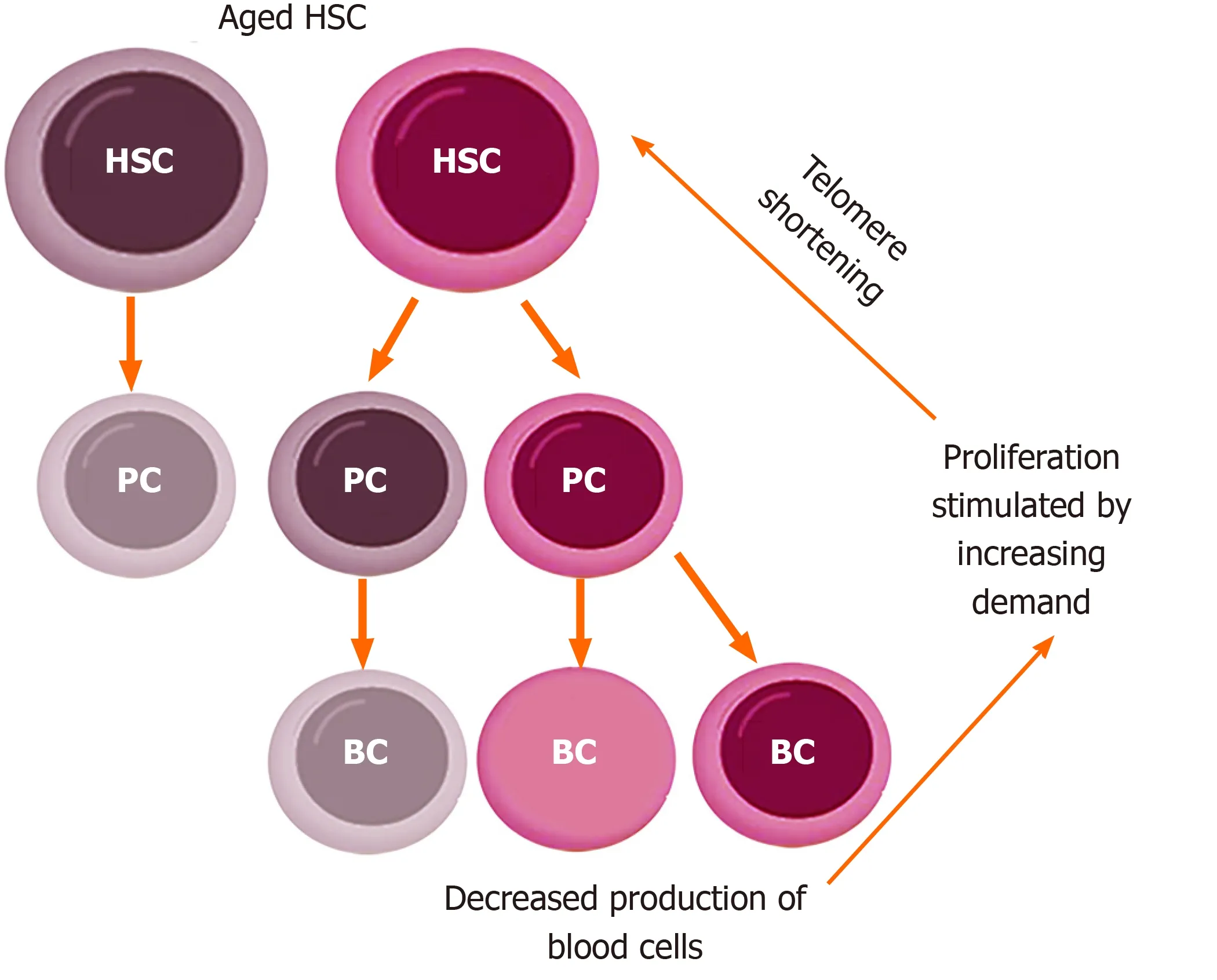
Figure 3 Model of increased proliferation in aging hematopoietic stem cells.
A specific kind of genomic damage is represented by dysfunctional telomeres.These are produced not only by telomere shortening (which we have discussed above), but also by generic DNA damage, which, if located at the telomeres, is less repairable[40].Dysfunctional telomeres permanently activate the DNA damage response pathway (γ-H2AX, p53, and p21)[40], which leads to a depletion of the HSC and progenitor poolsviaapoptosis and/or senescence[41,42].
Epigenetic changes
Besides accumulation of DNA damage, time-dependent epigenetic modifications seem to gradually lead to loss of HSC potential.This gradual loss of epigenetic marks during HSC replication and aging can explain the limited self-renewing capacity of HSC.Indeed, alterations of epigenetic writers or erasers severely affect HSC functionality[23].Moreover, the randomness of this process can explain the increased functional heterogeneity of HSC during aging observed in experimental studies in mice[23].
Several authors have investigated the changes of DNA methylation and histone modifications during HSC aging.Many lineage specific genes show high levels of euchromatin and H3K4me2, but are poorly expressed in HSC.This indicates that these genes are prepared to be activated in the differentiating progeny[43].Moreover, modifications in DNA methylation patterns during aging do not exert significant changes in HSC gene expression, but affect their progeny.For example, euchromatin regions comprising lymphoid genes likeBlnkandIrf8(which are not expressed in HSC, but are essential for B cell development) show increased DNA methylation as HSC age, and this leads to decreased lymphoid differentiation[44] (in agreement with the phenomenon of immunosenescence discussed above).On the other hand, HSC aging is accompanied by a decrease of the H3K4 demethylase KDM5B[45], hypomethylation of stem cell maintenance genes[46], and hypermethylation of transcription factors regulating HSC differentiation, such asPu.1[47].
In order to understand these combinations of DNA and histones methylation/ unmethylation, they can be imagined like the buttons play/pause/stop on a player (Figure 4).In young HSC, self-renewal genes are transcribed, while myeloid and lymphoid genes are “paused”[48].In the myeloid and lymphoid progeny, the respective genes are transcribed, while the others are repressed.In aged HSC, on the other hand, transcription of self-renewal genes is enhanced, myeloid genes are “paused” (thus ready to be expressed in the differentiating progeny), and lymphoid genes are repressed.
This indicate a picture where aging of HSC is characterized by epigenetic modifications that lead to an increase of self-renewal/expansion and a decrease of differentiating progeny.These features are also in agreement with the age-related prevalence and cellular features of myelodsysplastic syndromes.
Correlation and causality
It seems ascertained that HSC aging is correlated with the number of replications undergone[26].Cellular replication, in turn, causes phenomena that are linked to aging of HSC, including replication stress and telomere shortening.Moreover, a sustained rate of cellular division determines a higher cellular metabolism, which [through reactive oxygen species (ROS) production] could induce DNA damage.Therefore, it is not simple to find a link of causality that goes beyond simple correlation.
Several works discussed above demonstrated epigenetic modifications in aged HSC that mechanistically explain their features (increased self-renewal and myeloidskewed progeny).This epigenetic reprogramming, however, accurately describes an aged phenotype, but does not explain its causes.
Since HSC aging is linked to replication (which, in turn, is accompanied by loss of telomeric DNA), every epiphenomenon linked to aging will be correlated with telomere shortening.Nonetheless, the case of laboratory mice seems to show that it is not the decrease of telomere length that causesper seexhaustion of HSC.Several other causes (which we discussed above) like telomere dysfunction, epigenetic modifications, and accumulation of mutations seem the principal candidates of the aging phenotype of HSC.However, telomere shortening (without reaching lengths that induce senescence) could influence gene expressionviathe “telomere position effect”[49,50].It would be tempting to speculate that shortening of telomeres determines the epigenetic modifications discussed above, thus determining the aged phenotype.However, in our view, it is improbable that a highly conserved phenotype (HSC stemness, myeloid biased,etc.) could depend by a “telomere position effect” on genes whose location is different from one species to another.
As we have deduced that epigenetic modifications and telomere shortening are linked to HSC aging, but seem not be its causes, accumulation of DNA damage remains as a strong candidate.Accumulation of replication stress (DNA damage due to stalled replication fork) is clearly linked to the replicative history of a cell.Yanget al[51] and Pilzeckeret al[52] demonstrated that murine HSC have high levels of replicative bypass of DNA lesions, leading to DNA damage tolerance and the accumulation of DNA damage over time.However, human DNA repair systems are more efficient than murine ones[53], so it should be investigated if the high levels of replicative bypass of DNA lesions is present also in human HSC.
Besides replication stress, DNA damage can be the result of various insults, first of all presence of ROS.Indeed, in forcedly proliferating HSC, Walteret al[39] detected increased levels of ROS and 8-oxo-2’-deoxyguanosine.Moreover, DNA damage located on telomeres (which seems to be unrepairable, see above) could be a specific candidate, stochastically produced, but accumulating over time.Summing up, we presume that accumulation of DNA damage is the main causative factor determining the aged phenotype of HSC.
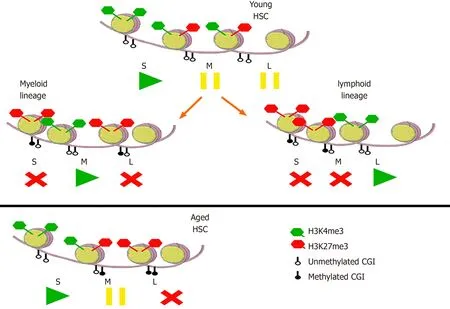
Figure 4 Epigenetic modifications in hematopoietic stem cells and their progeny.
CONCLUSION
Studies of the last 20 years have determined important advances in the understanding of HSC kinetics, growing and aging.Nonetheless, these new discoveries pose new questions, which future studies may solve.Are the switches between lymphoid- and myeloid-biased sub-populations of HSC present also in human infancy? At what time do they take place? What are the mechanisms and causal relationships between telomere shortening, DNA damage, epigenetic modifications, and HSC aging? Is a mechanism of telomere shortening induced by oxidative stress, and independent from replication, present in HSC? Answer to these questions may give further insights into the aetiology of pediatric leukaemias and myelodysplastic syndromes in the elderly, as well as a better comprehension of the hematopoietic system as a whole.
杂志排行

World Journal of Stem Cells的其它文章
- Genome engineering and disease modeling via programmable nucleases for insulin gene therapy; promises of CRISPR/Cas9 technology
- Immunotherapy in the treatment of lymphoma
- Recent trends in stem cell-based therapies and applications of artificial intelligence in regenerative medicine
- Epigenetic regulation of autophagy: A key modification in cancer cells and cancer stem cells
- Review of the potential of mesenchymal stem cells for the treatment of infectious diseases
- Therapeutic potential of periodontal ligament stem cells