Genome engineering and disease modeling via programmable nucleases for insulin gene therapy; promises of CRISPR/Cas9 technology
2021-07-24YunusEksiAhterSanliogluBaharAkkayaBilgeEsinOzturkSalihSanlioglu
Yunus E Eksi, Ahter D Sanlioglu, Bahar Akkaya, Bilge Esin Ozturk, Salih Sanlioglu
Yunus E Eksi, Ahter D Sanlioglu, Bahar Akkaya, Salih Sanlioglu, Department of Gene and Cell Therapy, Akdeniz University Faculty of Medicine, Antalya 07058, Turkey
Bilge Esin Ozturk, Department of Ophthalmology, University of Pittsburgh, Pittsburgh, PA 15213, United States
Abstract Targeted genome editing is a continually evolving technology employing programmable nucleases to specifically change, insert, or remove a genomic sequence of interest.These advanced molecular tools include meganucleases, zinc finger nucleases, transcription activator-like effector nucleases and RNA-guided engineered nucleases (RGENs), which create double-strand breaks at specific target sites in the genome, and repair DNA either by homologous recombination in the presence of donor DNA or via the error-prone non-homologous end-joining mechanism.A recently discovered group of RGENs known as CRISPR/Cas9 gene-editing systems allowed precise genome manipulation revealing a causal association between disease genotype and phenotype, without the need for the reengineering of the specific enzyme when targeting different sequences.CRISPR/Cas9 has been successfully employed as an ex vivo gene-editing tool in embryonic stem cells and patient-derived stem cells to understand pancreatic beta-cell development and function.RNA-guided nucleases also open the way for the generation of novel animal models for diabetes and allow testing the efficiency of various therapeutic approaches in diabetes, as summarized and exemplified in this manuscript.
Key Words: Programmable nucleases; CRISPR/Cas9; Stem cells; Disease modeling; Diabetes; Insulin gene therapy
INTRODUCTION
Approximately 25000 protein-coding and 25000 noncoding RNA genes totaling up to 50000 genes are estimated to exist over a meter of linear DNA in the human genome[1].In recent years, decoding the function of individual genes became the major goal of human genome research.Comprehensive genetic analysis was required to disclose correlations between genetic variants and human diseases.The development of effective therapeutic drugs against human genetic diseases depends on understanding the function of genes.Although the connection between genetic changes and human diseases has been known for years, the mutations that cause the emergence of some disease phenotypes can only be treated by genetic modification[2,3].Due to the complexity of the human genome, modification of genetic information represented a challenge requiring technologically advanced molecular tools[4,5].Furthermore, the efficient and safe delivery of these genome engineering tools to targeted tissues of interest remains to be a concern for the clinical application of these technologies[6].
The idea of genome modification was first introduced by Rudin and Haber[7] in 1988, where they reported efficient repair of homothallic switching endonuclease enzyme-triggered chromosomal breaks inSaccharomyces cerevisiae(S.cerevisiae) by homologous recombination (HR).They demonstrated that the efficiency of gene targeting is increasedviathe creation of double-strand DNA breaks (DSBs) in yeasts[7].Furthermore, the significance of HR in the repair of DSBs was first demonstrated in mammalian cells by Jasin’s research group[8].Previously, it was thought that lethal chromosomal DSBs in mammalian cells were repaired by a mechanism that does not require homology to the break site.This was a rather contrast withS.cerevisiaewhere the major DSB repair pathway is HR.To determine if mammalian cells used recombinational repair at a notable level, the DNA cleavage sites for the rare-cutting endonuclease I-SceI fromS.cerevisiaewere integrated into the mammalian (mouse) genome.Thus, specific DSBs could be introduced into the mouse genome at the enzyme’s cleavage sitesviaan expression system for the I-SceI.The use of a homologous DNA resulted in targeted genetic modification of mammalian cells mainly by HR or to a lesser degreeviaerror-prone non-homologous end joining (NHEJ) mechanism.These two studies not only demonstrated the importance of HR in the repair of DSBs in mammalian cells, but also laid the foundation for targeted genome modification.Yet it is really a challenge to reprogram the long DNA-binding recognition regions of the meganucleases for genomic modification.At this point, genome editingviaprogrammable nucleases constitutes a very efficient alternative approach to modify any desired region in the genome.
Gene targetingviaHR is not practical in higher eukaryotic cells due to low efficiency, hampering their routine use.On the other hand, programmable nucleases generating site-specific DSBs were found to increase HR efficiency by at least 100 fold and/or activated the error-prone NHEJ mechanism[9].Following genome editing approachesviazinc finger nucleases (ZFNs) that remained as the primary modification option for researchers for a while, transcription activator-like effector nucleases (TALENs) were developed as novel tools for genome editing, followed by the development of a new class of genome-editing nucleases named RNA-guided engineered nucleases (RGENs).RGENs which displayed their specificityviasmall guide RNAs, brought excitement to the field as easier to modify targeted nuclease systems.While each programmable nuclease has its exclusive properties, they all cleave the nuclear DNA at specific target sites as a similar mechanism of action, activating endogenous DNA repair systems leading to targeted genome modification.In this review, we would like to first describe the general features of programmable nucleases then specifically focus on RGENs for genome modification.
GENOME ENGINEERING WITH ZFNs AS TARGETABLE DNA CLEAVAGE REAGENTS
In the early 1990s, a biochemist named Srinivasan Chandrasegaran from Johns Hopkins University speculated and have shown that the type IIS restriction enzyme Fok1 had two separate protein domains, namely the DNA-binding domain and the domain that exerted the endonuclease activity, which could be separated from each other with the help of a protease, without losing function[10].This opened the way for the idea of combining the Fok1 nuclease site with DNA binding domains of other proteins to create sequence-specific nucleases, such as in the generation of ZFNs by binding of the Fok1 endonuclease to zinc finger proteins[11].Inspired by Chandrasegaran's work, Bibikovaet al[12] at the University of Utah succeeded inin vivogenome editing for the first time in a living organism by generating custom-designedDrosophilamutants by targeted cleavage using ZFNs.Although ZFNs use the nonspecific cleavage domain from the type II restriction endonuclease FokI as the cleavage domain, dimerization of FokI domains is required for cleavage which necessitates a pair of ZFNs targeting non-palindromic DNA sites (Figure 1)[13].Besides, directed evolution has been used to produce a FokI strain with enhanced cleavage activity[14].The cleavage specificity of FokI was also enhanced by modifying the dimerization interface using a structure-based design[15].
ZFNs are very useful to manipulate the genomes of plants, animals, and humans.They have successfully been used to correct disease-causing alleles in triplet repeat disorders, which are present in more than a dozen inherited neurological disorders including but not limited to Huntington's disease, myotonic dystrophy, and several spinocerebellar ataxias[16].Targeted delivery of the therapeutic genes to a preselected chromosomal locus can be achieved using plasmids encoding ZFNsviathe generation of DSBs in a specific gene in human cells.This would prevent all the complications associated with the viral delivery of therapeutic genes[17].Furthermore, patients' stem cells can be modified using ZFNsex vivo; after expanding them in culture, genetically modified stem cells can be put back into the patient to generate differentiated cells with corrected functions[18].
Unfortunately, many ZFNs have been shown to possess cytotoxic effects due to offtarget DSBs[19].Off-target cleavage activity of ZFNs is reported when the zinc finger domains are not specific enough for their target site or carry homology to other unintended sites.Generation of excessive amounts of DSBs may overwhelm the repair machinery leading to random integration of donor DNA and eventual cell death.To decrease off-target cleavage of 3-finger ZFNs that target two adjacent 9-basepair sites, the use of ZFNs with 4, 5, or 6 zinc fingers targeting longer and rare sites is suggested[20,21].Intriguingly, the competition between HR and NHEJ repair pathways represents a handicap for ZFN-mediated gene modifications.Inactivation of the catalytic activity of one ZFN monomer in the ZFN dimer results in the generation of Zinc-finger nickases (ZFNickases) which has shown to provide a bias for HR-mediated gene modification[22].ZFNickases possess a reduced spectrum of off-target alterations due to the reduction in NHEJ repairs.Unfortunately, the application of ZFNs to manipulate endogenous genes has been a challenging job due to difficulty in generating zinc finger domains that target the chosen sequence with sufficient sequence specificity.
TALENs IN TAILORED GENOME ENGINEERING
The solution to the cytotoxicity problem of ZFNs came in 2009 from naturally occurring transcription factors of a Gram-negative bacterial plant pathogenXanthomonas.Two research groups, one led by Bogdanoveet al[23] at Iowa State University and the other by Boch and Bonas[24] at Martin Luther University, revealed the interaction mechanisms betweenXanthomonas-originated transcription activatorlike effectors (TALEs) and DNA, and this led to the discovery that these proteins could also be used for genome editing with their simple DNA-binding code and relative ease of engineering.TALEs are proteins that are secreted byXanthomonasbacteria by way of type III secretion system following infection of plants.The DNA-binding domain of a TALE carries a repetitive highly conserved 33–34 amino acid sequence with divergent 12thand 13thamino acids referred to as the repeat variable Di-residue[25].These residues are highly flexible and used in specific nucleotide recognition.TALENs are generated by fusing a TAL effector DNA-binding domain to a DNA cleavage domain Fok1, which can be engineered to cleave specific sequences of DNA (Figure 1)[26].Although ZFNs and TALENs contain the same Fok1 nuclease domain at their Cterminal ends, their DNA binding sites are different from each other (Figure 1).Unlike zinc finger proteins, each repeat of TALEs recognizes a single base.
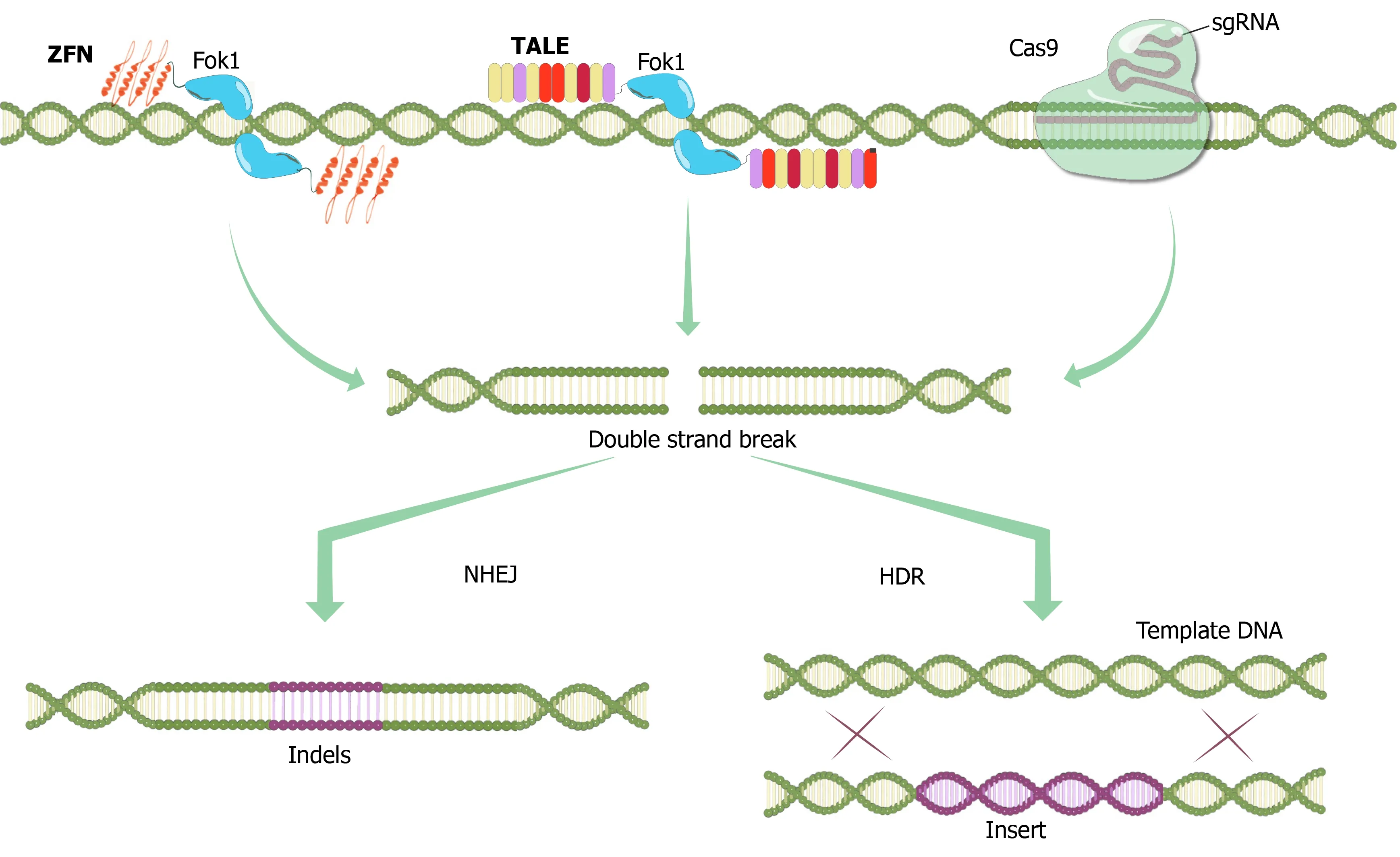
Figure 1 Programmable nucleases.
TALENs have been used to efficiently modify human embryonic stem cell (hESC) and induced pluripotent stem cell (iPSC) clones and human erythroid cell lines[27,28], to generate knockout mice and rats[29,30].TALENs have also been experimentally deployed to correct the genetic errors underlying diseases[31].The genetic defects that cause disorders such as sickle cell disease[28,32], xeroderma pigmentosum, and epidermolysis bullosa[33] were all amenable to correctionin vitrousing TALENs.Moreover, T cells can be genetically modified by TALENs to become resistant to chemotherapeutic drugs and display antitumor activity[34,35].
Unfortunately, the lack of an efficient delivery mechanism, immunogenicity, and the nonspecific TALEN binding to unintended sites limited thein situapplication of TALENs for the treatment of human diseases[31].Furthermore, the generation of the DNA segment containing the TALE repeat sequences is very difficult and timeconsuming.It is technically very hard to generate these sequences since they are likely to recombine with each other in cells.Although it is quite remarkable that TALENs showed minimal toxicity and off-targeting activity in human cells, the emergence of the CRISPR/Cas9 system has diverted much of the attention of the community to this new class of nucleases.
RNA-DEPENDENT GENOME MODIFICATION WITH CRISPR/CAS9; A NEW ERA OF GENETIC ENGINEERING
The studies that led to the discovery of clustered DNA repeats came independently from three different parts of the world.Ishinoet al[36] and his colleagues from Osaka University accidentally cloned part of a clustered regularly interspaced short palindromic repeats (CRISPR) sequence together with their gene of interest, the isozyme conversion of alkaline phosphatase gene, in 1987 without knowing the function of the interrupted clustered repeats.In 1993, Groenenet al[37] have discovered a cluster of interrupted direct repeats (DRs) while working on DNA polymorphism in the DR cluster ofMycobacterium tuberculosisin the Netherlands.Later, Mojicaet al[38] at the University of Alicante in Spain was studying the repeat sequences and their relevant function in the archaealHaloferaxandHaloarculaspecies when he observed the transcription of interrupted repeats for the first time in 2000.In 2001, while working on possible additional interrupted repeats, Mojica and Montoliu[39] suggested the use of the acronym CRISPR to prevent the misunderstanding curtailing from the numerous abbreviations utilized to define the sequences in the related literature.The evidence that showed some CRISPR spacers were derived from phage DNA and extrachromosomal DNA such as plasmids came from three separate research groups in 2005[40-42].CRISPR, which was discovered in the genomes of prokaryotic organisms like bacteria and archaea, is a family of DNA sequences derived from DNA segments of certain bacteriophages that had previously infected the prokaryote[43].CRISPR-associated protein 9 (Cas9) is an enzyme manifesting helicase and nuclease motifs that employs CRISPR sequences as a guide to identify and slice specific strands of DNA that are complementary to the CRISPR sequence.CRISPR sequences together with Cas9 enzymes established the foundation of a new technology known as CRISPR/Cas9 that is deployed to modify genes within organisms[44].
CRISPR/Cas systems with different properties identified from many bacteria and archaea species can be categorized into different groups.Class 1 systems use a complex of multiple Cas proteins to disrupt the target nucleic acids, while the Class 2 system exerts the same functionviaa single large Cas protein.Among these, the type II CRISPR/Cas9 system, which belongs toStreptococcus pyogenes (S.pyogenes)is the best known and the most commonly used technology as of today.This system functions as an immune system against viruses or foreign nucleic acids in bacteria[45].If the bacteria are infected with a phage, the phage DNA entering the bacteria causes the bacterial defense mechanisms to be activated.Many Cas proteins are synthesized from the bacterial CRISPR locus and take a piece (protospacer) from the phage DNA in an unknown mechanism, and add it together with a repeat sequence to the region separated by palindromic repeat sequences.Subsequently, transcription of the CRISPR locus that contains the pre-CRISPR RNA (crRNA), transactivator crRNA (tracrRNA) and Cas9 enzymes takes place[46].The pre-crRNA is processed to form the 20 nucleotide-long crRNA that is complementary to the viral DNA.Many Cas proteins are involved in this process.crRNA-tracrRNA and the Cas9 protein bind to the viral DNA by forming a complex.To bind to viral DNA, Cas enzymes specifically recognize the protospacer adjacent motif (PAM) sequences that must be present in the target DNA.These species-specific conserved sequences located near protospacers, matching the spacer sequences in CRISPR loci, were discoveredviacomputational analyses.For theS.pyogenesCas9 enzyme,this sequence is either "NAG" or "NGG", the latter being the most widely used PAM sequence in customized cleavage systems.
The Cas9 enzyme contains two nuclease activity domains.These are the HNH domain (creates a break in the complementary chain) and the RuvC domain (creates a break in the non-complementary chain).In the presence of the PAM sequence, the crRNA-tracrRNA-Cas9 complex binds to the viral DNA and creates double-strand breaks, thus prevents viral infection[47].
After this discovery, crRNA and tracrRNAs were combined without causing a decrease in Cas9 activity, thus the chimeric-hybrid guide RNA (sgRNA) was formed, also recognized by the Nobel Prize in Chemistry in 2020 as a very significant finding[48].Genome editing in human cell cultures using CRISPR/Cas9 was reported for the first time simultaneously by Hsuet al[49], Conget al[50] and Maliet al[51].Later, various CRISPR/Cas9 and sgRNA expression plasmids were reported.In this new system, the desired target gene could be modified with the Cas9 expression vector by designing a target-specific oligonucleotide of 20 nucleotides in length and transferring it to the sgRNA expression plasmid.
Potential off-target effects of the Cas enzymes, although the specificity of which is under strict control of the 20 nt guide sequence and the PAM region adjacent to the target sequence to be cleaved, are of major concern especially for therapeutic applications.Strategies to overcome this drawback include alteration of the sgRNA sequence for instanceviatruncation of the 3’ sequence, reducing the amount of the transfected DNA, or modifications in the Cas9 enzyme.In this context, a mutation was introduced to the 10thamino acid of the Cas9 enzyme, converting Aspartic acid to Alanine (D10A).This mutation caused the loss of RuvC nuclease activity of the Cas9 enzyme and enabled the creation of the nickase Cas9 (Cas9_D10A) mutant (Figure 2)[52].Nickase activity introduces only single-strand DNA breaks, which are repaired by a "base excision" repair mechanism that does not induce mutations in the DNA sequence.Furthermore, two different gRNAs are needed to create double-strand DNA breaks with nickase Cas9[53].Since it is very unlikely that two different sequences of 20 nucleotides exist in the same way in the genome, this makes Cas9_D10A-mediated DNA modifications safer[51].
After the nickase Cas9 protein, the ‘defective or dead Cas9’ (dCas9) protein was generated by silencing mutations in both the HNH and RuvC domains of the enzyme (D10A and H840A, respectively), with inactivated endonuclease activity thus retaining only the specific DNA-targeting function when guided by the sgRNA[52].The dCas9 protein is fused with many different proteins and used for various purposes such as transcriptional control as well as DNA modifications (Figure 2).Fusion of the Fok1 nuclease domain to the dCas9 protein reduced off-target effects.Researchers fused dCas9 with a series of tandem repeats (such as VP48, VP64, VP160) of the Herpes simplex viral protein 16 (VP16) trans-activation domain, together with sgRNAs designed to target the upper part of the transcription start site, thus activating transcription in the target gene (Figure 2)[54,55].Similarly, the fusion of a transcription-inhibiting protein blocks the target gene.The best defined and frequently used of these repressors is the Krüppel-associated box domain, a repressive chromatinmodifier domain, providing a 90%-99% knockdown of the target gene through heterochromatin spreading with minimal off-target effects[56,57].Moreover, fusing the dCas9 to epigenetic enzymatic domains (such as p300, SID4x, PRDM9, DOT1L, Tet1) enables specific epigenetic alterations, while base editing in specific DNA sequences can be initiatedviafusion of dCas9 with base editing enzymes (Figure 2)[56-59].Another CRISPR/Cas9 technology called CRISPR-genome organization offers a programmable 3D platform for studying the relationship between the nuclear structure and gene regulation and function (Figure 2)[60].
Of all the programmable nucleases available with unique properties used for various purposes, today the CRISPR/Cas9 system is considered the best choice for genome editing as an easy to apply, cost-effective, and versatile system.Moreover, effective gene edits in human tripronuclear zygotes using the CRISPR/Cas9 system were first described by Chinese scientists Lianget al[61] in 2015.A successful cleavage of mutant Beta-Hemoglobin in 28 out of 54 embryos was achieved using the CRISPR/Cas9 system.
In 2015, another type of nuclease, Cas12a (formerly known as Cpf1), was discovered from the bacteriumFrancisella novicida[62].Several key differences from Cas9 were observed in Cas12a such as requiring only a crRNA for successful targeting, relying on a T-rich PAM, and causing a staggered cut in double-stranded DNA.These features made Cas12a ideal for multiplexed genome editing.Also, Cas12a cleaves DNA 18–23 base pairs downstream from the PAM site without disrupting the recognition sequence after repair.
In 2016, a new RNA-guided RNA endonuclease, the nuclease Cas13a, was characterized from the bacteriumLeptotrichia shahii[63].Cas13a is directed by its crRNA to an ssRNA target and then cleaves other ssRNA molecules non-discriminately.This cleavage pattern of Cas13a called collateral cleavage has been exploited for the development of various diagnostic technologies[64-66].
Nonetheless, the CRISPR/Cas9 system, which functions as a bacterial defense system in nature, has been deployed in many areas of biotechnology as one of the most up-to-date gene modification methods today[67].
THE ROLE OF PROGRAMMABLE NUCLEASES IN PANCREATIC BETACELL DEVELOPMENT AND FUNCTION
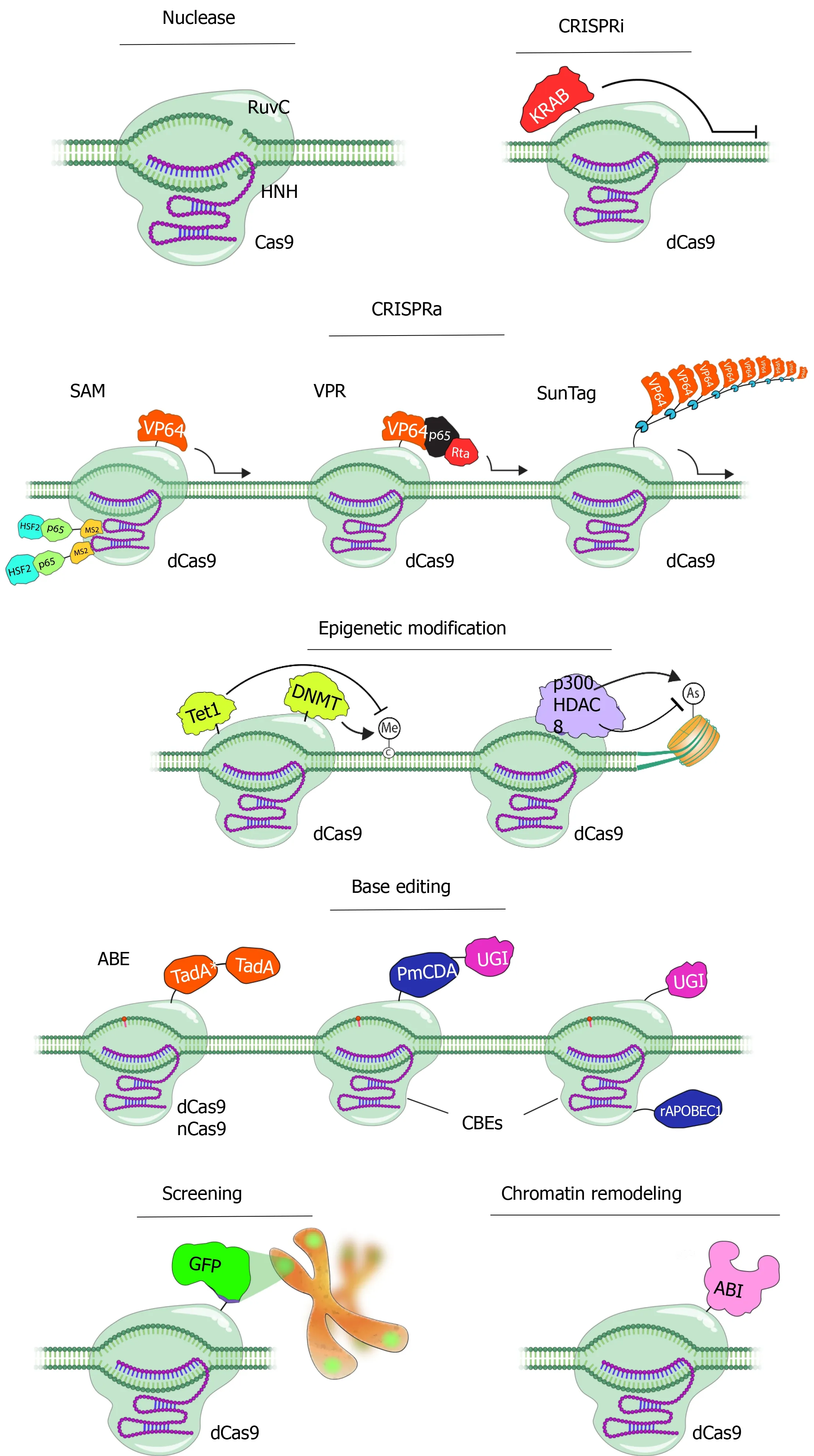
Figure 2 Applications of CRISPR/Cas9 technology.
The mechanisms of beta cell function in health and disease conditions can experimentally be dissected using CRISPR/Cas9 technologyviamanipulation of the genome in pancreatic beta cells[68,69].Moreover, the generation of novel genetically modified animal models for the testing of beta-cell function is now feasible using CRISPR/Cas9 technology.Pancreatic islet cell loss is one of the essential underlying causes of diabetes[70].As a solution to this problem, three different strategies have been employed for the recovery of pancreatic beta cells over the years.These three approaches are stimulation of the proliferation of existing beta cells, reprogramming various types of cells into beta cells, and inducing differentiation of beta cells from PSCs.Pancreatic islet-cell proliferation can also be inducedin vivousing gene therapy approaches as shown previously[71-73].In this context of utilizing pluripotent stem cells, pancreatic beta cell-like insulin-producing cells (IPCs) differentiated from ESCs, which were first isolated from human embryos in 1989; and iPSCs, which were first obtained by reprogramming of differentiated cells, have created excitement as new potential sources of beta cells[74-76].Ethical concerns, as well as the necessity of immune suppression, emerged in studies/potential treatment approaches related to ESCs led studies in this field focus mostly on iPSC-mediated approaches.Besides eliminating ethical concerns, this approach also facilitates disease modeling, drug development, and understanding of the pathogenesis of diabetes, paving the way for development of new-generation treatment strategies.Programmable nucleases (ZFNs, TALENs, CRISPR/Cas9) helped us understand the development, regeneration, insulin production, and secretion patterns of pancreatic beta cells in all aspects.
CRISPR/Cas9 system revealed species-specific differences in gene function.For an instance, McGrathet al[77] demonstrated that the lack of Neurogenin3 (NEUROG3), a basic helix-loop-helix transcription factor essential for endocrine pancreas formation in mice, did not interfere with the growth of the human endocrine pancreas.hESC lines carrying NEUROG3 knockout mutations efficiently formed endoderm and pancreatic progenitors but not the endocrine cellsin vitro.Furthermore, a 75%–90% knockdown of NEUROG3 expressionvialentivirus-mediated short hairpin RNA approach only reduced but did not prevent the pancreatic endocrine cell development, suggesting that although NEUROG3 is fundamental for human endocrine pancreas development, very little amounts are sufficient for pancreatic endocrine cell formation.
STAT3 has been shown to mediate the activation of Neurog3 in acinar cells reprogramming them into beta cells in diabetic mouse models[78].Besides, activating germline mutations in STAT3 were recently reported as a cause of neonatal diabetes mellitus associated with beta-cell autoimmunity[79].The investigation of the activating mutation, STAT3K392R, on pancreatic development using iPSCs derived from a patient with neonatal diabetes and pancreatic hypoplasia suggested that the STAT3K392Rmutation caused premature endocrine differentiation through direct induction of NEUROG3 expression.Fortunately, the disease phenotype was completely reversed using CRISPR/Cas9 technology to correct STAT3 mutation.
An efficient genome-editing platform in human PSCs (hPSCs) (iCRISPR) has recently been established through the use of TALENs and the CRISPR/Cas system[80].This approach combined the strength of genome editing and stem cell biology to methodically manipulate transcriptional control of pancreatic development and the developmental defects related to permanent neonatal diabetes mellitus[81].In this study, Zhuet al[82] silenced eight of the transcription factors (PDX1, RFX6, PTF1A, GLIS3, MNX1, NGN3, HES1, and ARX) effective in beta cell development in hESCs using CRISPR/Cas9 and TALENs.In addition to defining the specific developmental steps affected by these mutations, this approach uncovered new insights into disease mechanisms, concerning the role of RFX6 in controlling the number of pancreatic progenitors, a prerequisite for PDX1 in pancreatic endocrine development, and a potentially differing role of NGN3 in humans and mice.The deployment of programmable nucleases enabled the identification of transcription factors regulating pancreatic development, advancing hPSC-based beta cell replacement therapies for the treatment of diabetes.Although experimental animal models are needed for reproducing human diseases, genetic and metabolic differences between the species can sometimes cause failure to properly mimic human disease, preventing the development of an effective treatment.
The CRISPR/Cas9 system also significantly contributed to the evaluation of the results gathered from genome-wide association studies (GWAS).Although a large number of genes associated with type 2 diabetes (T2D) have been identified containing single nucleotide polymorphisms, insertions, deletions, and copy number variations in GWAS studies, making the connection between the pathophysiology of the disease and any possible use of the information in drug discovery is challenging[83-86].Utilizing CRISPR/Cas9, Zenget al[87] silenced three genes (CDKAL1, KCN111, KCNQ1) in hESC-derived glucose-responsive cells, which were identified by GWAS studies as susceptibility genes for T2D, and observed that although the silencing of these genes did not affect the generation of insulin+ cells, no change was detected in the beta-cell differentiation potential.However, insulin+ beta-cells were hypersensitive to glucolipotoxicity in the absence of the CDKAL1 gene expression.These preliminary studies have shown that functional properties of gene loci identified by GWAS with possible roles in the pathogenesis of diseases can be better assessed by utilizing CRISPR/Cas9 technology.
CONTRIBUTION OF PROGRAMMABLE NUCLEASES IN THE CREATION OF DISEASE MODELS FOR DIABETES
As isolated human islets are rare and valuable materials for diabetes research, hPSCs represent a good alternative to pancreatic islet cell donors.iPSCs can be established from adult somatic cellsviadirect reprogramming and differentiated into beta cell-like insulin-producing cells[88].Although full maturation of these cells into IPCs that secrete insulin in response to changes in blood glucose concentration cannot be achieved easily, this approach can be used to model developmental defects of genetic diseases like diabetes[68].Genome editing mediated by CRISPR/Cas9 technology provides strong opportunities to examine the effects of specific genotypes and to develop disease models with genetic changes that cause diabetic syndromes, and likely to rapidly increase the possibilities for studying beta cell physiology[89].
Type 1 diabetes (T1D) results from the destruction of the pancreatic beta cells by the autoimmune system due to the impairment of multiple immune tolerance mechanisms[90].T1D in humans is strongly associated with an allelic variant of protein tyrosine phosphatase nonreceptor type 22 (PTPN22), PTPN22R620W, the existence of which increases the risk of diabetes by two- to fourfold[91].Since the NOD mouse is a spontaneous T1D model sharing many diabetes-related genetic pathways with humans, the introduction of the murine orthologous PTPNR619Wmutation to the NOD genome was expected to enhance the spontaneous development of T1D.Microinjection of CRISPR/Cas9 and a homology-directed repair template into NOD single-cell zygotes resulted in mice with PTPNR619Wmutation exhibiting increased insulin autoantibodies and earlier onset and higher penetrance of T1D[92].
Leptin receptor (Lepr) functions as a receptor for the fat cell-specific hormone leptin (ob), which regulates energy homeostasis, the balance between food intake and energy expenditure[93].The Lepr-defective mice (db/db) is currently the most widely used mouse model of T2D, exhibiting severe obesity, hyperphagia, polydipsia, and polyuria[94].The rat equivalent of the db/db mouse is the Zucker rat (fa/fa), which has a spontaneous autosomal mutation in the Lepr gene and exhibits a comparable phenotype of hyperphagia leading to glucose intolerance, insulin resistance, and morbid obesity[95].Intriguingly, the Zucker rat does not exhibit hyperglycemia.To solve this, the Lepr knockout rats were generated using the CRISPR/Cas9 system[96].These Lepr CRISPR/Cas9 knockout rats exhibited all the diabetes-related phenotypes expanding animal models for biomedical and pharmacological research on obesity and T2D.Besides, some problems observed in leptin receptor-deficient db/db mouse and Zucker rat animal models concerning persistent hyperglycemia and late appearance of glucose intolerance were overcome using CRISPR/Cas9-generated Lepr knockout rats.
Although rodent models are useful for studying diabetes, the use of animal models metabolically closer to humans is a more effective way to understand the development and pathogenesis of human diseases using the CRISPR/Cas9 system.Despite diabetes research can benefit from using pigs as a model owing to sharing similar physiology and metabolic pathways to humans, the scarcity of pig models exhibiting diabetes symptoms represents a clear handicap.For this purpose, Choet al[97] first modified the insulin gene in somatic cells using the CRISPR/Cas9 system.Then, somatic cell nuclear transfer carrying the modified insulin gene resulted in the generation of pig embryos with insulin (INS) knockout phenotype.INS knockout piglets manifested hyperglycemia and glucosuria demonstrating the effectiveness of CRISPR/Cas9-mediated generation of novel pig models, which might be more suitable for drug development and islet transplantation studies compared to rodents.
Also, the CRISPR/Cas9 system can be an effective way to develop disease models of monogenic diabetes such as Maturity Onset Diabetes of the Young (MODY) referring to any of the several hereditary forms of diabetes mellitus caused by mutations interrupting insulin production[98].Despite MODY is an autosomal dominant disease, which requires only one abnormal gene to manifest the disease symptoms, the severity of the disease is regulated by the presence of a second allele.While MODY 2 and MODY 3 are the most common forms, mutations in the insulin gene cause MODY10, which is often confused with T2D owing to the similarities in the clinical symptoms[99].Although the course of the disease is mild, people with this genetic disorder require insulin use in the following years.Such single gene disorders, which cannot be treated with current treatment methods, can be effectively treated with gene therapy.Here, lentivirus-mediated insulin gene therapy may be a permanent solution for MODY10 treatment as shown previously for T1D[72,100].
A STRATEGY TO CREATE A CRISPR/CAS9-MEDIATED INSULINDEFICIENT PANCREATIC BETA-CELL LINE SUITABLE FOR TESTING THE THERAPEUTIC EFFICACY OF INSULIN GENE DELIVERY VEHICLES
Although a certain reduction in the pancreatic beta cell mass is achieved in streptozotocin (STZ)-induced experimental diabetic animal models, beta cells that remain alive and capable of synthesizing and secreting insulin may interfere with the testing of insulin gene therapy strategies.To fully test the efficacy of insulin gene therapy vectors, pancreatic beta cells that do not synthesize insulin are needed.Since these cells are not commercially available, CRISPR/Cas9 system provides a suitable method to generate pancreatic beta cells without insulin synthesis.The insulin gene can be silenced at the DNA level with the help of the CRISPR/Cas9 using specific guide RNAs targeting the insulin gene.Since these cells will have all beta-cell characteristics except for insulin synthesis, it will be a suitable model for bothex vivoandin vivotesting of the efficacy of insulin gene transfer vectors.To create this model, pancreatic beta cells are transfected with a silencing plasmid encoding the CRISPR Cas9 protein, specific guide RNA, and a HR plasmid containing the homologous regions to the insulin gene (Figure 3).In this strategy, the silencing plasmid creates a double-strand break in the insulin gene, while the HR plasmid delivers an antibiotic resistance gene to selectively purify successful transformants, and a fluorescent protein-encoding gene (e.g.RFP) to confirm successful recombination in the region where the DSB occurs.Thus, a pancreatic beta-cell line is generated with stable fluorescent protein expression, that can be visually examined under a fluorescent microscope after transfection.These cells can be purified by flow cytometry due to the stable fluorescent protein expression they contain, or by antibiotic selection with the transferred antibiotic resistance gene (Figure 3).Then, the cells are cloned using the limited dilution method.Finally, the insulin secretion status of single-cell colonies can be confirmed by insulin ELISA, by western blot, or immunocytochemistry methods.After confirmation, the insulin gene can be deliveredin situby gene therapy vectors to determine if insulin secretion is restored following the transduction of INS knockout pancreatic beta cells.Then, INS knockout beta cells generated by CRISPR/Cas9 technology with or without insulin gene delivery can be transplanted under the kidney capsule of STZ-induced diabetic animals (Figure 4).By doing so, a pancreatic beta-cell line deficient in insulin gene synthesis can be generated and can be used forin vivotesting of the efficacy of insulin gene therapy vectors.

Figure 3 Production of CRISPR/Cas9-mediated insulin-deficient pancreatic beta cell line for testing the therapeutic efficacy of insulin gene therapy vectors.
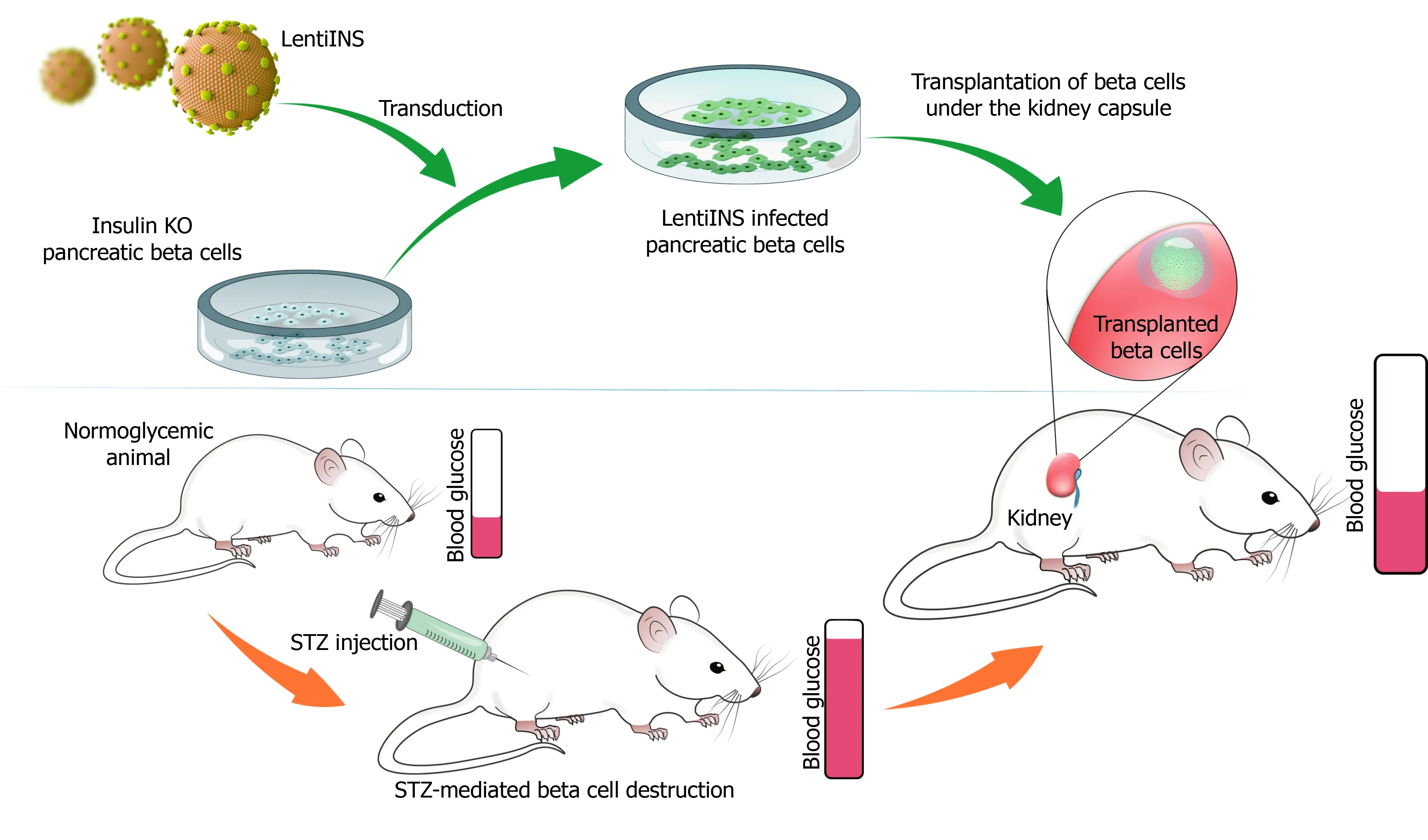
Figure 4 Testing the therapeutic efficacy of the lentivirus vector carrying insulin gene (LentiINS).
CONCLUSION
In summary, the CRISPR/Cas9 system is a unique technology that provides a very powerful and versatile platform for genome editing, with ease of design and implementation, high efficiency, and low cost[67].Described as a game-changer in genetic engineering, one of the main purposes of using the CRISPR/Cas9 technology is to develop suitable models for drug testing and to understand the molecular and physiological mechanisms underlying the development of human diseases[18].Among the diseases for which successful cell-based CRISPR/Cas9-mediated disease models were generated are cystic fibrosis, Barth syndrome, β-thalassemia, Duchenne muscular dystrophy, and hemophilia A[101-106].Mouse models for tyrosinemia and lung cancer, and rat and primate models for muscular dystrophy have also been successfully created with the CRISPR/Cas9 technology, with many more that are worked on and to be developed[107-110].As an additional approach, as described in this publication, a newin vivodiabetes disease model can be developed by allogenic transfer of an INS knockout pancreatic beta-cell line that is created by CRISPR/Cas9 technology under the kidney capsule of STZ-induced diabetic animals.To determine the therapeutic efficacy of insulin gene therapy, the same procedure needs to be repeated with insulin-deficient pancreatic beta cells only after the insulin gene delivery.With the current advances in the powerful CRISPR/Cas9 technology, there is a better chance for overcoming the challenge of generating and implementing the most accurate, specific, and predictive disease models to provide a better understanding and treatment of human diseases.
杂志排行

World Journal of Stem Cells的其它文章
- Immunotherapy in the treatment of lymphoma
- Recent trends in stem cell-based therapies and applications of artificial intelligence in regenerative medicine
- Epigenetic regulation of autophagy: A key modification in cancer cells and cancer stem cells
- Review of the potential of mesenchymal stem cells for the treatment of infectious diseases
- Growing and aging of hematopoietic stem cells
- Therapeutic potential of periodontal ligament stem cells