RORA基因截短变异致智力发育障碍伴小脑共济失调一家系的临床分析与基因诊断
2021-07-23刘舒张也韦思思欧阳海梅梁金群陈暖曾伟宏江剑辉
刘舒 张也 韦思思 欧阳海梅 梁金群 陈暖 曾伟宏 江剑辉
(1.广东省妇幼保健院 儿童遗传代谢与内分泌科,广东 广州 511442;2.广东省妇幼保健院医务科,广东 广州 511442)
智力发育障碍伴小脑共济失调(intellectual developmental disorder with cerebellar ataxia)是一种常染色体显性遗传病,目前报道的文献非常少,它是由于染色体15q22.2区域的RAR相关孤儿受体A(RAR related orphan receptor A,RORA)基因变异引起的生长发育障碍(OMIM#600825)[1]。RORA基因变异可以导致多种异常临床表现,包括智力发育障碍伴小脑共济失调、发育迟缓、行走能力延迟、语言发育迟缓、斜视、远视、癫痫发作、隐性脊柱裂、共济失调、肌张力减低、运动延迟、眼球震颤、慢性便秘、边缘性人格障碍、动眼运动异常、智力残疾、脑桥小脑萎缩、胼胝体发育不良、眼睑肌阵挛、小脑发育不全、全面发育迟缓、自闭症行为、弱视、锥体功能障碍、震颤、肾积水等[1,2]。本病的智力发育障碍和步态失调呈进行性发展,如果能够早期确诊,对患儿的临床症状缓解和后期的康复结局起到非常重要的作用。目前RORA基因变异导致智力发育障碍伴小脑共济失调的发病机制已基本明确,但因临床表现复杂多样,导致诊断和鉴别诊断非常困难。本研究采用家系全外显子测序发现了RORA基因新的致病位点,扩展了该病的基因型与临床表型谱。
1 材料与方法
1.1 临床资料 患儿男孩,2020年11月首次到广东省妇幼保健院儿童遗传代谢与内分泌科门诊就诊,主诉为:出生后发现生长发育落后,以运动发育迟缓为著。首次就诊时年龄为4岁3个月,临床表现为行走不稳,不能独走,步态异常,易跌倒,语言表达差,无有意义发音,对外界刺激无反应,手势表达少。门诊拟诊“精神运动语言发育迟滞,共济失调”收入本院儿科病房。入院后对患儿进行相关检查。患儿男孩,2016年9月出生,其母亲产前检查一切正常,G1P1,孕40周顺产。患儿出生体重3.0kg,出生时无窒息,Apgar评分10分,喂养正常。运动发育史:7个月开始爬,22个月会站立,25个月开始扶走,走路不稳,摇摇晃晃,且经常摔倒;语言发育史:无有意义发音,对外界刺激毫无反应,手势表达少。父母籍贯广东广州,非近亲婚配;否认家族遗传病史;患儿有一弟弟,2018年2月出生,生长发育正常。目前患儿大小便不能自理,听力正常,咽反射基本正常。眼科检查:无视神经萎缩迹象;神经系统检查:步态异常,行走不稳,摇摇晃晃,易摔跤;共济运动检查:指鼻试验(-),跟膝胫试验(-),闭目直立试验(+)。四肢肌力低下,膝反射(+),腹壁反射(+),双侧自发巴氏征(+),颈无抵抗,布氏征(-),克氏征(-)。
1.2 辅助检查
1.2.1 常规检查 患儿入院后除了进行心肌酶谱、肝肾功能、电解质、血常规等检查外,还进行血糖、血氨、铜蓝蛋白、微量元素、血气分析、铁蛋白、血脂、甲状腺功能等代谢相关检查;同时进行了颅脑的影像学检查;以及遗传代谢病血串联质谱和尿液气相质谱检测。
1.2.2 全外显子组基因检测 本研究通过了本院医学伦理委员会审查批准,征得患儿父母的知情同意。抽取患儿静脉血5ml,采用Blood DNA Kit V2 CW2553血液提取试剂盒提取血液DNA,运用第二代测序技术(next generation sequencing,NGS)进行分析。DNA全基因组文库的制备:利用KAPA Library Preparation Kit (Illumina ® platforms): KR0453-v3.13取750ng DNA样本进行超声波打断,得到150~200 bp的DNA片段。将片段末端补平后用AMPure XP 纯化磁珠纯化,纯化后的DNA片段两端加A碱基(A-Tailing)并连接接头,连接后产物进行8个循环的PCR扩增。DNA样本捕获:在Agilent SureSelectXT2 Target Enrich System反应系统内对真空浓缩DNA文库样本进行杂交捕获。杂交混合液在65℃环境下孵育24h。而后加入Dynabeads MyOne Streptavidin T1 (Invitrogen)。杂交捕获后的产物进行PCR扩增,13循环。PCR产物采用Agencourt AMPure XP 纯化磁珠进行纯化并用Qubit dsDNA HS Assay Kit进行定量。高通量测序:取捕获后的DNA样本进行Illumina novaseq高通量测序。测序数据经Illumina Sequence Control Software(SCS)评估合格后,进行数据读取和生物信息学分析。
1.2.3 测序数据分析及Sanger测序验证 变异位点致病性评级及数据解读规则参考美国医学遗传学和基因组学会(American College of Medical Genetics and Genomics,ACMG)指南[3]及ClinGen序列变异解释(sequence variant interpretation,SVI)专家组对指南标准的应用建议[4-6],排除千人基因组、 ExAC、 gnomAD等数据库中突变频率大于1%的变异位点,去除非功能性变异位点(如同义突变、非编码区突变等),再经过致病性预测(SIFT、Polyphen2、CADD等软件)、临床症状对照、相关疾病数据库查询与文献参考等综合考虑,找到候选基因变异位点进行家系验证。所用注释数据库为: Human Genome38 (hg38/GRCh38)、 RefSeq、 dbSNP、 1000 Genomes phase3、 ExAC、 gnomAD等数据库,所用解读数据库包括DGV、 DECIPHER、 OMIM、 UCSC、 ClinVar、 HGMD及PubMed等数据库。随后根据WES中发现的变异分别设计引物,进行PCR扩增以进行Sanger测序,验证全外显子组测序(whole exome sequencing,WES)的结果及遗传模式。测序结果通过MutationSurveyor软件进行序列比对分析,并进行家系验证。
2 结果
2.1 临床结果
2.1.1 常规检查结果 患儿常规生化检查和血液、尿液的串联质谱检查显示结果无明显异常,排除常见氨基酸、有机酸和脂肪酸代谢异常相关遗传代谢性疾病。
2.1.2 影像检查结果 影像学检查:颅脑MRI平扫显示双侧脑室稍增宽,双侧小脑半球体积减小,脑沟加深、增宽,提示轻度脑积水和小脑萎缩(图1)。
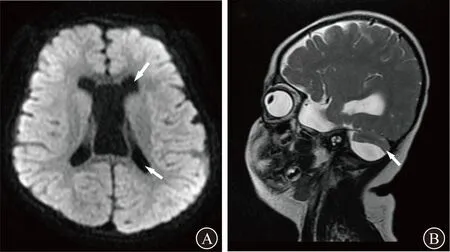
图1 患儿颅脑MRI检查
2.2 全外显子组基因测序及Sanger验证 测序结果发现一个与患儿临床表型高度相关的基因RORA:c.79C>T (p.R27*)(NM_002943.3)杂合截短变异位点。此变异位于RORA基因第1号外显子区域,该变异使第 27位密码子 CGA 转变为TGA,导致精氨酸(R)转变为终止密码子,即 p.R27*。50名家系外正常对照和父母DNA测序未发现相应变异,提示该变异可能为新生致病性变异(图2B、2C)。
2.3 新变异的致病性分析RORA基因c.79C>T (p.R27*)(NM_002943.3)变异在两种功能预测软件中都提示有害(SIFT Score:0.001,MutationTaster Score:1)。
2.4 新变异蛋白的结构域分析 新生变异c.79C>T位于RORA蛋白N末端,使该蛋白发生提前终止 (p.R27*),导致RORA蛋白缺失522个氨基酸肽链(图2A)。经过生物信息学功能预测,RORA蛋白提前终止,极有可能影响该蛋白的生物学功能(图2D、2E)。

图2 患儿家庭RORA基因变异测序结果和生物信息学分析
3 讨论
基因变异位点RORA:c.79C>T (p.R27*)(NM_002943.3)在一般人群数据库gnomAD中未收录,该变异尚未在HGMD数据库收录,为新发现的变异位点。该截短变异在两种功能预测软件中都提示有害,根据ACMG指南,该变异评级为疑似致病变异。根据患儿的临床表现和基因分析结果,此患儿诊断为常染色体显性智力发育障碍伴小脑共济失调。RORA基因新生变异致病有报道称表型特征为发育迟缓、行走能力延迟、语言发育迟缓、斜视、远视、癫痫发作、隐性脊柱裂、共济失调、肌张力减低、运动延迟、眼球震颤、慢性便秘、边缘性人格障碍、动眼运动异常、智力残疾、脑桥小脑萎缩、胼胝体发育不良、眼睑肌阵挛、小脑发育不全、全面发育迟缓、自闭症行为、弱视、锥体功能障碍、震颤、肾积水[1,2]。本例患者变异与该病已经报道过的表型有相似的特征;头颅磁共振成像(magnetic resonance imaging,MRI)显示双侧小脑半球体积减小,脑沟增宽、加深,说明有小脑萎缩。本例患儿的原因可能与RORA基因变异使运动神经元受损相关。尽管RORA基因变异已经在一些神经退行性疾病的动物模型中被描述过[7-9],但是儿童患病报道非常少[1],说明对于RORA基因导致常染色体显性智力发育障碍伴小脑共济失调的表型程度和致病机制的研究和认识还非常缺乏。因此,我们这例患儿发病的确切机制需要进一步的功能研究来阐明不同的RORA基因变异所带来的临床表现差异。
RORA基因位于15q22.2,全长约741kb,包含11个外显子[10,11]。迄今为止,共发现18种变异,均为复杂重排。本研究的家系发现的c.79C>T (p.R27*)为新生截短变异,该变异使RORA蛋白发生提前终止(p.R27*),导致RORA蛋白缺失522个氨基酸肽链。经过生物信息学功能预测,RORA蛋白提前终止,极有可能影响该蛋白的生物学功能。该变异尚未见报道,也不是已知的多态性改变,很可能是一个新的致病性变异位点,但尚需进行功能分析证实。
本研究首次在一常染色体显性智力发育障碍伴小脑共济失调患儿发现RORA基因c.79C>T (p.R27*)杂合变异,且该变异为新生变异。我们的研究明确了患儿发病的遗传学病因,为患儿的精准治疗和后续该家庭的产前诊断和优生优育,打下了坚实的基础。
