基于网络相似性整合算法的lncRNA与miRNA相互作用预测研究
2021-07-23丁尉哲吴卷书刘宏生
丁尉哲,南 洋,吴卷书,崔 迪,张 力,2,3,刘宏生,2,3
(1.辽宁大学生命科学院;2.辽宁省生物大分子计算模拟与信息处理工程技术研究中心;3.辽宁省药物分子模拟与设计工程实验室,辽宁 沈阳 110036)
lncRNA是一类长度超过200个核苷酸的非编码RNA,可通过多种机制发挥其生物学功能[1]。miRNA是生物体内源基因编码的长度约为21~25个核苷酸的非编码小RNA,主要通过与靶mRNA互补配对在转录水平上对基因的表达进行负调控。越来越多研究表明,lncRNA与miRNA的相互作用也共同控制着一些复杂疾病的生理过程[2-3]。目前已有多种计算方法用于lncRNA-蛋白质相互作用和lncRNA-疾病相互作用预测,但只有少数几个模型能用于预测lncRNA-miRNA相互作用[4]。本研究基于lncRNA和miRNA的序列、功能和表达谱数据及已知的lncRNA-miRNA互作数据,通过网络相似性整合算法,预测lncRNA与miRNA互作对。该研究为lncRNA-miRNA互作预测建立了新的方法,同时也为疾病机理分析提供了新思路。
1 材料与方法
1.1 材料
1.1.1 lncRNA与miRNA关联数据 本研究中lncRNA与miRNA相互作用数据来源于lncRNASNP2数据库(http://bioinfo.life.hust.edu.cn/lncRNASNP#!/)[5]。在删除无效和重复的记录后,共下载了10597条互作数据,其中包括780种不同类型的lncRNA和275种不同类型的miRNA。
1.1.2 lncRNA和miRNA的序列信息 lncRNA的序列信息来自LNCipedia数据库(https://Incipedia.org/)[6]。miRNA的序列信息来自miRBase数据库(http://www.mirbase.org/index.shtml)[7]。在删除重复的记录及无效的序列信息之后,共获得7263个相互作用对。此外,为了更好地验证本研究方法的有效性,进一步考虑了表达谱相似性和功能相似性信息。lncRNA的表达谱信息和功能注释信息来自NONCODE数据库(http://www.noncode.org/)[8]。去除无效和冗余信息后获得了用于计算表达谱的449个lncRNA和用于计算功能注释的264个lncRNA。此外,在删除无效和重复的信息后,在microRNA数据库(http://www.mirbase.org/)中获得了230个miRNA的表达谱信息,并在miRTarBase 7.0数据库(http://miRTarBase.mbc.nctu.edu.tw)中获得272个miRNA的功能数据[9]。
1.2 方法
1.2.1 lncRNA序列相似性的测定 使用基于字符串匹配的Needleman-Wunsch算法[10]对lncRNA进行序列比对,并将lncRNA序列相似性结果归一化到0到1的范围内,其中1对应于两个lncRNA完全相同,而0则对应于缺少与该序列相关的相似性信息。具体公式如下:
(1)
其中LR(li,lj)为lncRNAli和lj之间的序列相似性,ns(li,lj)为lncRNA中两个比较序列之间的最大匹配数。
1.2.2 miRNA序列相似性的测定 miRNA序列之间相似度可由下列公式计算:
(2)
其中MR(mi,mj)为miRNAmi和mj之间的序列相似性,ns(mi,mj)为miRNA中两个比较序列之间的最大匹配数。
1.2.3 预测lncRNA与miRNA关联数据 通过引入邻接矩阵Y可以更好地描述lncRNA-miRNA的关系。若lncRNAli与miRNAmj相互作用,则矩阵元素Yij为1,否则为0,其中字母l和m分别代表实验中涉及的lncRNA和miRNA。同时,序列相似性数据中矩阵Y的维数被设为780×275,而利用表达谱相似性数据和功能相似性数据得到的矩阵维数则分别为449×230和264×272。
1.2.4 NSILMI方法预测lncRNA与miRNA相互作用 本研究提出通过整合miRNA和lncRNA的向量空间得分来计算潜在的lncRNA-miRNA互作得分的方法,命名为:NSILMI(Network Similarity Integration Method for predicting LncRNA-MiRNA Interactions,NSILMI)。在本研究中,主要采用余弦相似度来计算向量空间得分。
在lncRNA向量空间中,向量VLRi被用来描述lncRNAi与所有lncRNA之间的相似性并由LRi(矩阵LR的第i行)来表示。同样地,向量VYj被用来描述miRNAj和所有lncRNA之间的相似性并由Yj(矩阵Y的第j列)进行表示
VLRi=LRiVLRi=LRi
(3)
VYj=YjVYj=Yj
(4)
lncRNA空间分数被定义为:
(5)
其中VLRi·VYjVLRi·VYj是向量VLRi和VYj的点积,||VLRi||是向量VLRi的范数,||VYj||是向量VYj的范数,而NSILMI_L(i,j)则为向量VLRi和VYj的余弦相似度。当VLRi和VYj之间的夹角越小时,向量空间分数NSILMI_L(i,j)就越大。
因此,在lncRNA-lncRNA相似网络中,与lncRNAi相关的lncRNA空间相似性越高,lncRNAi与miRNAj之间的关联相似度就越大。同样地,与miRNAj相关的lncRNAs的空间相似性越高,lncRNAi与miRNAj之间的关联相似度也就越大。
而在miRNA向量空间中,向量VMRj被用来表示miRNAj与所有miRNAs之间的相似性并由MRj(矩阵MR的第j列)来表示它。向量VYi则被用来表示lncRNAi与所有miRNAs之间的相似性并由Yi(矩阵Y的第i行)表示
VLRj=LRj
(6)
VYi=Yi
(7)
MiRNA空间分数被定义为
(8)
其中VYi·VMRj是向量VMRj和VYi的点积;||VMRj||是向量VMRj的范数,||VYi||是向量VYi的范数,而NSILMI_M(i,j)则为向量VMRj和VYi的余弦相似度。当VMRj和VYi之间的夹角越小时,向量空间得分NSILMI_M(i,j)就越大。
所以,在lncRNA-miRNA相似网络中,与lncRNAi相关的miRNAs空间相似性越高,lncRNAi和miRNAj的关联相似度就越大。同样地,与miRNAj相关的miRNAs的空间相似性越高,lncRNAi与miRNAj的关联相似度越大。
1.2.5 整体打分 将lncRNA空间分数和miRNA空间分数集成为
NSILMA(i,j)=α×NSILMA_L(i,j)+(1-α)×NSILMA_M(i,j)
(9)
其中,α是平衡两个空间相似性的贡献的参数,α∈(0,1) ,NSILMI(i,j)是lncRNA i对miRNA j的预测相关分数。
1.2.6 模型评估 本研究采用100次重复的5折交叉验证检验NSILMI的准确性。采用ROC (receiver operating characteristic,ROC)曲线和ROC曲线下面积(area under curve,AUC)作为性能指标来评价NSILMI的预测准确性。一般情况下,AUC值在0.5和1.0之间,AUC值越大,模型越好[11]。
TPR(True Positive Rate)为分类器预测的阳性样本中真阳性样本的比例,同时也是ROC曲线的x坐标(以Sensitivity表示)。FPR(False Positive Rate)则表示阳性样本占分类器预测的阴性样本的比例,同时也是ROC曲线的纵坐标(以1-specificity表示)。在本研究中,使用如下公式进行计算
(10)
(11)
其中FP、TP、FN和TN分别是假阳性、真阳性、假阴性和真阴性。之后采用Logistic回归分类器,将每个样本概率设为正值并通过界定临界值(0.6)将大于或等于0.6的概率为阳性,小于0.6的概率为阴性。然后通过计算一组FPR和TPR可以得到平面上相应的坐标点。最终,随着临界值的逐渐降低,越来越多的样本归为阳性,但这些阳性也与真阴性混在一起,即TPR和FPR会同时增加。而当临界值最大时,对应的坐标点为(0,0)。相反,当临界值最小时,对应的坐标点则为(1,1)。
2 结果
2.1 整体打分结果为了找到合适的值,通过实验考察了从0.1到1的不同值。图1显示,当为0.7时,基于序列的NSILMI获得了最高的预测性能。
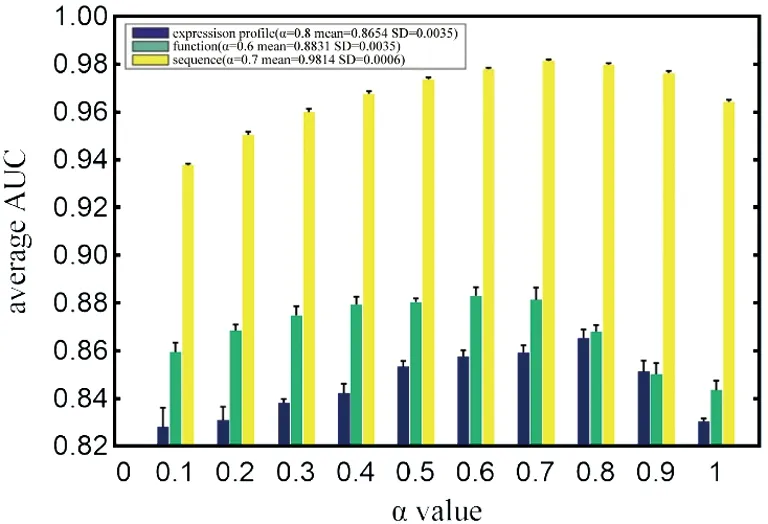
图1 不同α值下NSILMI的平均AUC
2.2 NSILMI与文献中报道模型的比较为了测试NSILMI模型的预测准确性,通过计算AUC值对NSILMI和其他网络预测方法的质量进行了评估。在本研究中,NSILMI与其他五种方法:LMFNRLMI[12]、NDALMA、LMI-INGI[13]、KATZLDA[14]和CF[15]进行了比较。在这些网络预测方法中,CF是常用的协同过滤算法。通过挖掘lncRNA已知互作数据,CF能够预测出与lncRNA显著相关的miRNAs,并根据不同的参数对lncRNA组进行划分,以便推荐具有相似参数选择的miRNAs。KATZLDA算法则可以合成已知关系来构建异构网络。其主要利用lncRNA-miRNA关联矩阵构建组合网络,并计算综合异构网络中每个节点的数目和长度,从而推断出lncRNA-miRNA对的得分。
图2展示了五种方法计算的AUC值。由图2可知,NSILMI获得了更好的AUC值(0.9814),高于LMFNRLMI (0.9554)、NDALMA (0.9255)、LMI-INGI (0.8917)、KATZLDA (0.8253)和CF (0.6713)。因此,与其他方法相比,NSILMI方法在推断潜在的lncRNA-miRNA相互作用方面拥有更高的精度。
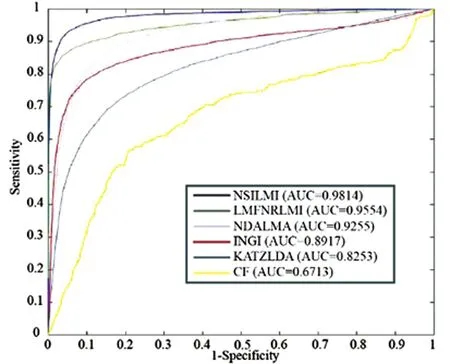
图2 NSILMI与LMFNRLMI、NDALMA、LMI-INGI、KATZLDA和CF预测性能的比较
2.3 不同类型相似度比较为了进一步验证NSILMI的有效性和适用性,我们分别使用基于表达谱相似度和基于功能相似度的数据作为测试集。为了更直观地比较不同类型相似度计算的效果,图3展示了AUC值的比较结果。
如图3所示,基于功能相似度和表达谱相似度计算的AUC值分别为0.8831和0.8654并低于基于序列相似度的0.9814。因此,虽然基于表达谱相似度和功能相似度的AUC值低于基于序列相似度的AUC值,但是结果仍都在0.85以上,这充分证明了我们方法的适用性和优越性。
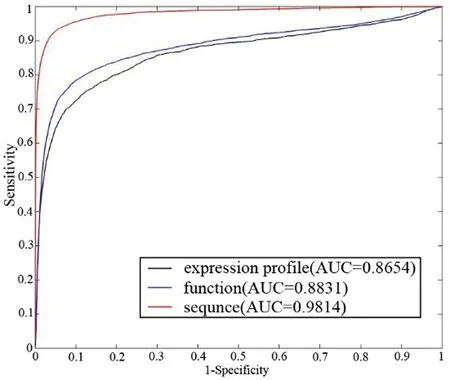
图3 基于不同相似度的NSILMI预测性能的比较
2.4 实例测试为了评估了NSILMI模型预测未知lncRNA-miRNA相互作用的能力,使用lncRNASNP2数据库的更新版本预测了新的lncRNA-miRNA互作对。表1列出了由NSILMI模型预测并排名前20个的lncRNA-miRNA互作对,其中16个互作对已被实验证实存在。

表1 NSILMI预测的前20个新互作对及其在预测中的排名
3 讨论
先前基于miRNAs的研究已经证明,miRNAs参与了人类疾病的病理过程并可以作为某些疾病的标志物。其中,miR-193b-3p的下调可能通过介导NCAPG的上调来促进胃癌细胞的增殖[16]。这些结果证明了NSILMI模型在预测lncRNA-miRNA相互作用的实用性。尽管仍有部分未被实验证实,但随着实验文献和数据库的完善,将会有越来越多的lncRNA-miRNA互作对得到证实。
lncRNA和miRNA在细胞的生理调控中发挥了至关重要的作用。其中,lncRNA DILC可以在胆囊癌组织中高水平表达,并且与生存预后有关[17]。而膀胱癌组织中miR-212的相互作用则与HMGA2密切相关,并能通过靶向调控HMGA2影响膀胱癌细胞的增殖、迁移和侵袭能力[18]。
而lncRNA和miRNA的相互作用在一些复杂疾病中也参与了重要作用。其中LncRNA UCA1通过胶质瘤中miR-206/CLOCk轴促进了细胞的生长和侵袭,因此LncRNA UCA1/miR- 206/CLOCk轴可能是神经胶质瘤的一个潜在的新靶点[19]。此外,lncRNAs HIF1A-AS2则通过与miR-153-3p结合来促进HIF-1α的上调,从而促进缺氧状态下HUVEC中的血管生成[20]。因此,lncRNA和miRNA的相互作用被认为是一些复杂疾病的重要原因。
本研究利用lncRNA和miRNA的序列、功能和表达谱信息,以及已知的lncRNA-miRNA相互作用信息,采用网络相似性整合算法,建立了可靠的lncRNA-miRNA互作预测模型。通过与其他网络预测方法比较,证明了NSILMI模型在推断潜在的lncRNA-miRNA互作对具有较好的效果。同时,我们也利用表达谱相似度和功能相似度验证了该模型的适用度并取得了较好的结果。这些结果均表明NSILMI模型在预测lncRNA-miRNA相互作用上具有可靠性和适用性。
同时,虽然上述实验取得了较好的效果,但基于本模型的预测仍存在一定的局限性。首先,由于该算法依赖于已被实验测定的相互作用数据,导致样本缺乏,可能会导致过拟合,因此需要使用了更多的实验数据进行训练和学习。其次,因为模型的参数选择非常复杂,很难得到完美的组合来提高预测精度。最后,本研究所使用的表达谱相似度、功能相似度、序列相似度,相似度矩阵的计算依赖于大量的计算资源。因此,在实际应用中,相似度的计算会给在线预测工具的开发带来困难。
综上所述,我们利用lncRNA和miRNA相互作用数据建立了NSILMI,成功对lncRNA-miRNA互作对进行预测。该项研究为lncRNA-miRNA关联的预测提供了新手段,也为复杂疾病的研究提供新方法和思路。
