无乳链球菌和海豚链球菌早期预警分子检测
2021-07-21黎晶晶张辉杰许德麟张其中
崔 淼,吴 敏,刘 茹,黎晶晶,张辉杰,许德麟,张其中
( 暨南大学 水生生物研究所,热带亚热带水生态工程教育部工程研究中心,广东省高校水体富营养化与赤潮防治重点实验室,广东 广州 510632 )
罗非鱼(Oreochromis)是世界范围内广泛养殖的水产品之一,据统计,2014年中国罗非鱼养殖产量已达到1.6985×106t,约达全球罗非鱼养殖总产量的32%[1]。近年来,罗非鱼链球菌病日益严重,给罗非鱼养殖业造成严重的经济损失。研究表明,罗非鱼链球菌病的主要致病菌是海豚链球菌(Streptococcusiniae)和无乳链球菌(S.agalactiae)[2]。无乳链球菌是一种人、畜、鱼共患的致病菌,可引起新生儿患脑膜炎、败血症等[3]。海豚链球菌至少能够感染27种海洋和淡水鱼类,死亡率高达30%~50%,而且能通过病鱼感染人类,属机会性人畜共患病原菌[4]。罗非鱼链球菌病的优势菌群会发生改变,2006—2007年间,海豚链球菌是罗非鱼链球菌病的主要病原菌,占总检出率的94.7%[5]。2008年至今,罗非鱼链球菌病主要由无乳链球菌引起,而海豚链球菌仍然是罗非鱼的潜在养殖风险[6]。海豚链球菌和无乳链球菌均能造成罗非鱼的严重病害,现有的防治方法除抗生素外还包括中草药和疫苗等[7-8]。但仅从外部症状和两种病原菌的菌落、菌体形态上,都很难准确判定病害是由哪一种链球菌引起的[9],这将严重影响到对链球菌病害及时、有效的处理。
目前罗非鱼链球菌的常规诊断方法有电镜观察、生化反应和药敏试验等[10],但这些方法均很难对感染鱼样本进行早期分子诊断和预警,同时一些生理反应现象可能随外界环境的变化而改变,从而容易导致误判情况的发生[11]。普通PCR每次仅能检测1个基因,可能会造成漏检或误检[12];多克隆抗体和单克隆抗体都是需要提前准备的,而且荧光抗体和ELISA的价格高且操作难[13-14];尽管等温环介导(LAMP)较方便,但容易出现假阳性现象[15];荧光定量PCR(qPCR)技术的操作要求较高,而且设备较为昂贵[16]。因此,为达到对罗非鱼链球菌的早期预警,需要建立更加快速、高效的早期分子预警检测技术。无乳链球菌与海豚链球菌的16S rRNA基因的同源性高度相似,达到96.6%以上[17],但是可在16S rRNA基因的变异区设计特异性引物,对两种菌进行区分和鉴定。表面免疫源性蛋白(Sip)属于编码B群链球菌的表面蛋白基因类群[18],在不同种的血清型菌株中均有存在[19]。笔者选用无乳链球菌的Sip基因和海豚链球菌16S rRNA基因的特异性序列,设计引物并对引物组合优化,建立了检测较低含量的无乳链球菌和海豚链球菌的双重PCR方法,旨在为罗非鱼链球菌的早期预警提供一种灵敏、特异的分子检测技术。
1 材料与方法
1.1 材料
1.1.1 菌株
无乳链球菌标准菌株ATCC13813、海豚链球菌标准菌株ATCC29178为中国水产科学研究院珠江水产研究所惠赠。无乳链球菌的分离菌株和海豚链球菌的分离菌株来源于罗非鱼病鱼,由本实验室分离、鉴定和保存;嗜水气单胞菌(Aeromonashydrophila)、迟钝爱德华氏菌(Edwardsiellatarda)、维氏气单胞菌(A.veronii)、创伤弧菌(Vibriovulnificus)、温和气单胞菌(A.sobria)、溶藻弧菌(V.alginolyticus)、大肠杆菌(Escherichiacoli)等菌株,均由本实验室分离、鉴定和保存。
1.1.2 试验鱼
试验鱼为尼罗罗非鱼(O.niloticus),体质量(50±5) g,由广东省罗非鱼良种场提供。实验室驯养14 d,确认正常无异样后可用于试验。
1.1.3 主要试剂
三羟甲基氨基甲烷、十二烷基苯磺酸钠、无核酸酶水、1×TE缓冲液、溶菌酶、蛋白酶K、苯酚/氯仿/异戊醇、无水乙醇、70%乙醇;dNTP(25 mmol/L)、10×PCR Buffer(无Mg2+)、MgCl2(25 mmol/L)、无菌超纯水、rTaq DNA聚合酶(5 U/μL);脑心浸液培养基、琼脂粉。
1.2 方法
1.2.1 引物的设计与合成
根据GenBank中海豚链球菌的16S rRNA基因部分序列,设计引物P1(5′-ATACCGCATAAGAGTGATT-3′)和P2(5′-ACCACCTGTCACTTCTGCT-3′),预期扩增片段大小为614 bp;根据无乳链球菌特异性基因Sip的序列,设计引物P3(5′-TGAATAAGAAATTGTTTTTAGCG-3′)和P4(5′-ACTTGTAATCGTTGGTTCTGCC-3′),预期扩增片段大小为870 bp,由华大基因有限公司合成。
1.2.2 DNA模板的提取
从-80 ℃冰箱保存的菌种中,挑取海豚链球菌标准株、无乳链球菌标准株、嗜水气单胞菌、迟钝爱德华氏菌、维氏气单胞菌、创伤弧菌、温和气单胞菌、溶藻弧菌、大肠杆菌。在无菌条件下接种于脑心浸液琼脂培养基上,在28 ℃恒温培养24 h后收集菌体,各细菌基因组DNA的提取步骤,参考革兰氏阴性菌和革兰氏阳性菌DNA的提取方法[20]。
1.2.3 双重PCR反应体系的优化
双重PCR引物组合和温度条件的优化为:用ddH2O分别将16S rRNA基因引物和Sip基因引物稀释为10 μmol/L,然后取16S rRNA上、下游引物[(0.5 μL,0.5 μL)、(0.4 μL,0.4 μL)、(0.3 μL,0.3 μL)],依次命名为A1、A2、A3;再取Sip基因上、下游引物[(0.5 μL,0.5 μL)、(0.4 μL,0.4 μL)、(0.3 μL,0.3 μL)],依次命名为B1、B2、B3。通过AxBy(x=1,2,3;y=1,2,3)进行组合(表1)。

表1 无乳链球菌和海豚链球菌引物比例试验组合
反应体系为:10×PCR Buffer (无Mg2+) 2.5 μL,MgCl2(25 mmol/L) 1.5 μL,dNTP (25 mmol/L) 2.5 μL,rTaq DNA 聚合酶 (5 U/μL) 0.1 μL,引物为表1中的AxBy(x=1,2,3;y=1,2,3),模板各l μL,ddH2O补充至25 μL。反应条件为:95 ℃预变性5 min;95 ℃变性30 s,52、54、56、58 ℃退火30 s,72 ℃延伸30 s,扩增30个循环;72 ℃延伸10 min。
反应完成后,取不同退火温度条件下每个引物组合的PCR扩增产物5 μL,与1 μL 6×Loading Buffer充分混匀,在恒压120 V、1.2%的琼脂糖凝胶下电泳20 min。如果16S rRNA和Sip基因两个目的条带同时出现,且无其他杂带,其对应的引物组合和退火温度为最佳条件。并以此建立双重PCR检测无乳链球菌和海豚链球菌的方法。
1.2.4 双重PCR特异性的测定
分别取无乳链球菌、海豚链球菌、嗜水气单胞菌、迟钝爱德华氏菌、维氏气单胞菌、创伤弧菌、温和气单胞菌、溶藻弧菌、大肠杆菌的基因组DNA和无乳链球菌和海豚链球菌基因组DNA的混合样本,每种模板为1 μL。根据筛选出的最佳反应条件进行PCR扩增,然后在恒压120 V、1.2%的琼脂糖凝胶下电泳20 min。
1.2.5 双重PCR灵敏度的测定
DNA含量的测定为:将无乳链球菌基因组DNA 10倍梯度稀释为9.84×10-2、9.84×10-3、9.84×10-4、9.84×10-5、9.84×10-6ng/μL;海豚链球菌基因组DNA 10倍梯度稀释为9.30×10-2、9.30×10-3、9.30×10-4、9.30×10-5、9.30×10-6ng/μL。根据筛选出的最佳反应条件和反应体系进行扩增,以能检测到的2种细菌基因组DNA的最大稀释质量浓度,确定双重PCR的DNA灵敏度。
1.2.6 双重PCR可重复性的检测
取无乳链球菌和海豚链球菌各5株,提取其基因组DNA,方法同上。以2种菌基因组DNA混合样本为模板,根据筛选出的最佳反应条件和反应体系进行PCR扩增。重复检测3次,均以标准菌株ATCC13813和ATCC29178的DNA混合样本为模板,作为阳性对照,确定双重PCR方法的可重复性。
1.2.7 双重PCR对模拟临床样品的检测
各取150 μL菌液密度为1.48×107cfu/mL的无乳链球菌、1.20×107cfu/mL的海豚链球菌,采用腹腔注射法同时人工感染尼罗罗非鱼18尾,24 h后取其肾脏组织。用十二烷基苯磺酸钠法结合溶菌酶法[21],提取肾组织内细菌基因组DNA。再用所建立的双重PCR方法进行检测,以确定双重PCR的临床适用性。
2 结果与分析
2.1 优化后的引物组合和退火温度
由图1可见,将16S rRNA和Sip基因引物的9种组合进行PCR扩增后,通过对引物组合进行相同反应体系和反应条件下的优化选择,A1B2引物扩增在52、54、56、58 ℃时比其他8种引物组合条带更清晰且特异性更高,因此选择A1B2作为引物扩增。即16S rRNA上、下游引物(10 μmol/L)的体积为0.5、0.5 μL,Sip基因上、下游引物(10 μmol/L)的体积为0.4、0.4 μL。在相同的反应体系下,退火温度为54 ℃时的双重PCR的特异性条带比52 ℃的条带清晰,且退火温度为54 ℃时的双重PCR条带比56、58 ℃的条带特异性高,所以最终选择54 ℃作为最佳的退火温度。
利用优化后的反应体系和反应条件,对无乳链球菌和海豚链球菌标准菌株的混合样本进行检测,出现两条特异性条带;用单一引物分别扩增16S rRNA和Sip基因,均出现1条特异性条带,且条带位置与预期结果一致(图2)。将特异性条带序列分别与无乳链球菌16S rRNA和海豚链球菌Sip基因序列BLAST比对,结果表明,本研究建立的双重PCR方法扩增出的特异性条带,为无乳链球菌16S rRNA和海豚链球菌Sip基因目的片段。
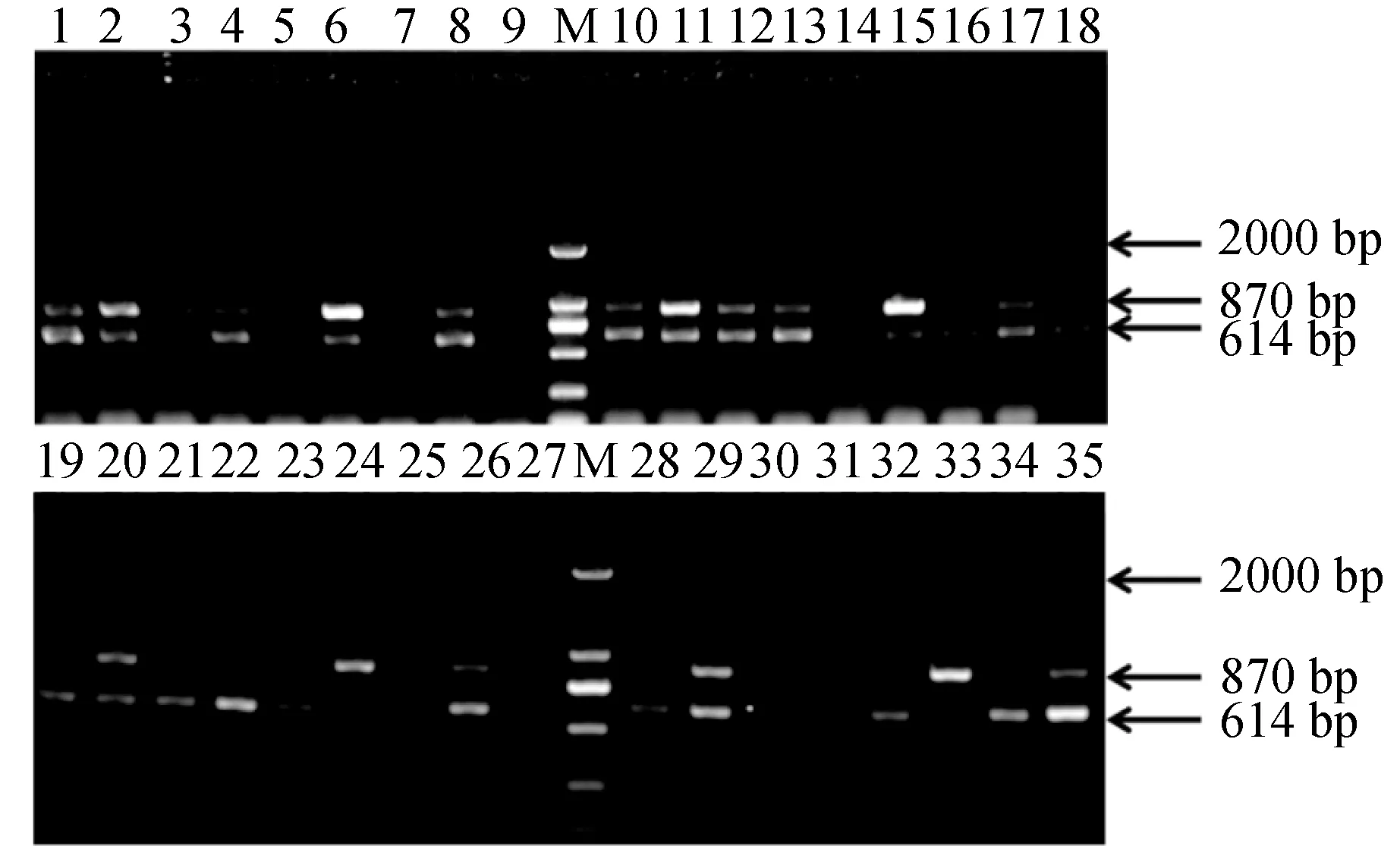
图1 双重PCR的引物组合优化结果Fig.1 Primer combination optimization of duplex PCRM.2000 bp DNA标记; 1~9.退火温度为52 ℃时,引物组合1~9的PCR扩增结果; 10~18.退火温度为54 ℃时,引物组合1~9的PCR扩增结果; 19~27.退火温度为56 ℃时,引物组合1~9的PCR扩增结果; 28~35.退火温度为58 ℃时,引物组合1~8的PCR扩增结果.M.2000 bp DNA marker; 1—9.PCR amplification results of primer combinations 1—9 at the annealing temperature of 52 ℃; 10—18.PCR amplification results of primer combinations 1—9 at annealing temperature of 54 ℃; 19—27.PCR amplification results of primer combinations 1—9 at annealing temperature of 56 ℃; 28—35.PCR amplification results of primer combinations 1—8 at annealing temperature of 58 ℃.
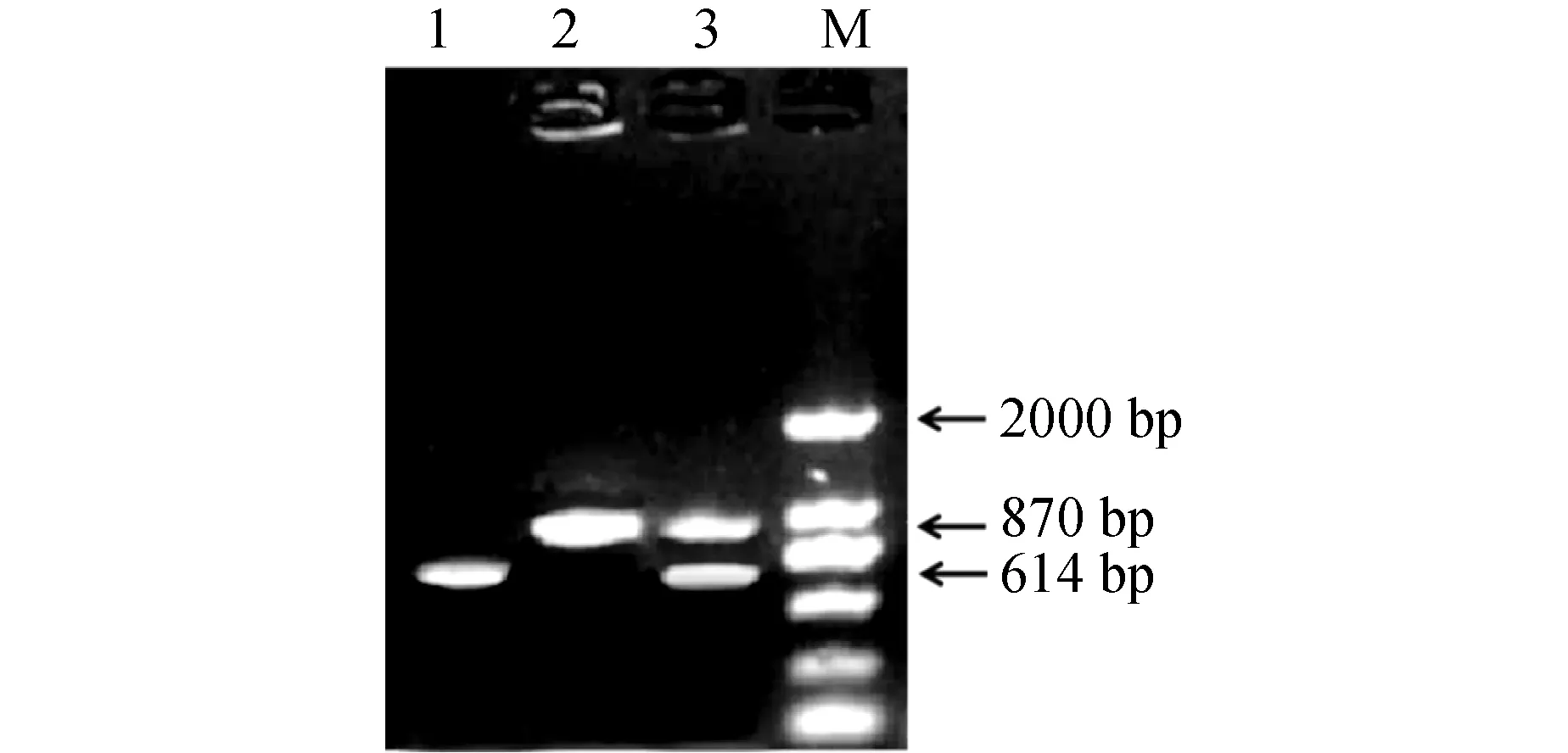
图2 单一引物和两对引物PCR扩增结果Fig.2 PCR amplification of single primer and two pairs of primersM.2000 bp DNA标记; 1.Sip基因; 2.16S rRNA基因; 3.双重PCR的结果.M.2000 bp DNA marker; 1.Sip gene; 2.16S rRNA gene; 3.the result of duplex PCR.
2.2 双重PCR检测无乳链球菌和海豚链球菌的特异性
由图3可见,无乳链球菌基因组DNA样品扩增的条带为870 bp;海豚链球菌基因组DNA扩增的条带为614 bp;两种菌的基因组DNA混合样品可同时扩增出870 bp和614 bp的目的条带;以嗜水气单胞菌、迟钝爱德华氏菌、维氏气单胞菌、创伤弧菌、温和气单胞菌、溶藻弧菌、大肠杆菌的基因组DNA为模板进行扩增,均未出现任何条带。

图3 无乳链球菌和海豚链球菌双重PCR特异性扩增结果Fig.3 Specific amplification of duplex PCR in S. agalactiae and S. iniaeM.2000 bp DNA标记; 1.无乳链球菌基因组DNA; 2.海豚链球菌基因组DNA; 3.无乳链球菌和海豚链球菌基因组DNA混合样品; 4~10.分别为嗜水气单胞菌、迟钝爱德华氏菌、维氏气单胞菌、创伤弧菌、温和气单胞菌、溶藻弧菌、大肠杆菌的基因组DNA.M.2000 bp DNA marker; 1 .the genomic DNA of S. agalactiae; 2.the genomic DNA of S. iniae; 3.the mixed genomic DNA of S. agalactiae and S. iniae; 4—10.the genomic DNA of A. hydrophila, E. tarda, A. vickers, V. vulnificus, A. temperate, V. alginolyticus and E. coli, respectively.
2.3 双重PCR检测无乳链球菌和海豚链球菌的灵敏度
用本研究建立的双重PCR方法,分别扩增不同质量浓度的无乳链球菌和海豚链球菌基因组DNA混合样本(图4)。分别扩增不同质量浓度的无乳链球菌和海豚链球菌的混合菌液(图5)。检测结果表明,该体系检测无乳链球菌和海豚链球菌的灵敏度均较高,其能检测到的无乳链球菌基因组DNA最低质量浓度为9.84×10-5ng/μL,海豚链球菌基因组DNA最低质量浓度为9.30×10-5ng/μL,无乳链球菌菌液最低密度为2.76×103cfu/mL,海豚链球菌菌液最低密度为2.51×103cfu/mL,两种菌的最低检测密度均低于半致死密度107cfu/mL,可用于罗非鱼链球菌的早期预警分子检测。

图4 无乳链球菌和海豚链球菌双重PCR 的DNA敏感性扩增结果Fig.4 DNA sensitivity amplification of duplex PCR in S. agalactiae and S. iniae M.2000 bp DNA标记; N.阴性对照; 1~5.分别为无乳链球菌DNA质量浓度9.84×10-2、9.84×10-3、9.84×10-4、9.84×10-5、9.84×10-6 ng/μL和海豚链球菌DNA质量浓度9.30×10-2、9.30×10-3、9.30×10-4、9.30×10-5、9.30×10-6 ng/μL.M. 2000 bp DNA marker; N. negative control; 1—5.the DNA concentration of 9.84×10-2 ng/μL, 9.84×10-3 ng/μL, 9.84×10-4 ng/μL, 9.84×10-5 ng/μL, and 9.84×10-6 ng/μL in S. agalactiae, and the DNA concentration of 9.30×10-2 ng/μL, 9.30×10-3 ng/μL, 9.30×10-4 ng/μL, 9.30×10-5 ng/μL, and 9.30×10-6 ng/μL in S. iniae, respectively.

图5 无乳链球菌和海豚链球菌双重PCR菌落灵敏度扩增结果Fig.5 Results of sensitivity amplification of S. agalactiae and S. iniae by duplex PCRM.2000 bp DNA标记; N.阴性对照; 1~5.分别为无乳链球菌菌液密度2.76×106、2.76×105、2.76×104、2.76×103、2.76×102 cfu/mL和海豚链球菌菌液密度2.51×106、2.51×105、2.51×104、2.51×103、2.51×102 cfu/mL.M.2000 bp DNA marker; N.negative control; 1—5.the bacterial solution concentration of 2.76×106 cfu/mL, 2.76×105 cfu/mL, 2.76×104 cfu/mL, 2.76×103 cfu/mL, and 2.76×102 cfu/mL in S. agalactiae, and the bacterial solution concentration of 2.51×106 cfu/mL, 2.51×105 cfu/mL, 2.51×104 cfu/mL, 2.51×103 cfu/mL, and 2.51×102 cfu/mL in S. iniae, respectively.
2.4 双重PCR检测无乳链球菌和海豚链球菌的可重复性
各取5株无乳链球菌和海豚链球菌,以2种菌的基因组DNA混合样本为模板,在优化的双重PCR反应体系下,均能同时扩增出2条特异性条带,与阳性对照的扩增结果相同(图6)。重复性检测3次的结果完全一致,说明该检测方法具有良好的重复性。

图6 重复性检测结果Fig.6 The results of repeatability testM.2000 bp DNA标记; 1~5.无乳链球菌和海豚链球DNA混合样本为模板; 6.阳性对照.M.2000 bp DNA marker; 1—5. a mixed sample of S. agalactiae and S. iniae DNA as template; 6.a positive control.
2.5 双重PCR检测模拟临床样品的结果
取18尾同时人工感染无乳链球菌和海豚链球菌的罗非鱼肾脏样本,提取组织内的细菌基因组DNA,以其作为模板进行双重PCR反应,能同时扩增出870 bp和614 bp的特异性条带,阳性检出率为100%(图7)。

图7 双重PCR对同时人工感染无乳链球菌和海豚链球菌的罗非鱼肾组织检测Fig.7 Detection of kidney in tilapia infected artificially with S. agalactiae and S. iniae by duplex PCRM.2000 bp DNA标记; 1~18.18个同时人工感染无乳链球菌和海豚链球菌的罗非鱼肾中提取的细菌DNA为模板.M.2000 bp DNA marker; 1—18.18 bacterial DNA extracted from tilapia kidney artificially infected with S. agalactiae and S. iniae at the same time.
3 讨 论
链球菌给全球鱼类养殖造成每年约2.5亿美元的重大经济损失,其中罗非鱼影响最大,其链球菌病的主要病原菌是无乳链球菌和海豚链球菌[22-25]。因此,研究出快速、高效、灵敏的早期预警分子检测技术,对实现罗非鱼链球菌的早发现、早防御和早治疗具有重要的经济意义和社会价值。
3.1 链球菌早期预警的特异性
本研究建立的分子检测技术为双重PCR,其是在普通PCR的基础上改进的,具有灵敏、经济、简便的优点。该检测体系通过一个PCR反应,能同时快速检测或鉴定两种病原微生物[26],节省了时间、试剂和费用,可广泛应用于水产病原菌的检测。在双重PCR反应中,引物的设计要考虑较多的因素[27],本研究结合了引物的特异性、扩增效率、引物二聚体等因素,获得了两对较为理想的引物。其中16S rRNA引物是根据海豚链球菌异于无乳链球菌的16S rRNA基因特异性序列设计的,克隆得到的16S rRNA基因片段由可变区和恒定区交叉排列构成,可以体现出这两种菌的差异,也可以测序得到目标序列。另一Sip基因引物是在无乳链球菌Sip基因特异性序列上设计而成,Sip基因编码了表面免疫源性蛋白,是B群链球菌表面源性蛋白的特异性基因,因此,利用Sip基因引物可以克隆得到无乳链球菌的特异性片段。这两对引物能分别扩增出无乳链球菌和海豚链球菌的特异基因片段,而无重叠和其他任何杂带,并且目的片段的大小能够在琼脂糖凝胶电泳中明显区分。人工同时感染无乳链球菌和海豚链球菌,罗非鱼组织样品的阳性检出率为100%,与细菌分离鉴定的结果相同,由此可知,该检测方法也具有较高的特异性。
3.2 链球菌早期预警的灵敏性
关于罗非鱼链球菌的分子检测技术也有报道,如通过巢式PCR鉴定出无乳链球菌[28];黎炯等[29]利用无乳链球菌的cfb基因和海豚链球菌16S rRNA基因序列建立的双重PCR,其检测灵敏度分别为无乳链球菌3.2×10-3ng/μL和海豚链球菌3.0×10-2ng/μL。而本研究建立的双重PCR可以检测到:无乳链球菌基因组DNA质量浓度为9.84×10-5ng/μL,海豚链球菌基因组DNA质量浓度为9.30×10-5ng/μL,检测到的目标菌基因组含量更低,灵敏度更高。Rodkhum等[30]根据海豚链球菌的lctO基因和无乳链球菌的16S rRNA基因建立的双重PCR,在目标菌密度为106cfu/mL的鱼组织中可以检测到该菌株。而本研究建立的双重PCR可以检测到:无乳链球菌菌液密度为2.76×103cfu/mL,海豚链球菌菌液密度为2.51×103cfu/mL,更低于目标菌的半致死密度107cfu/mL,适用于罗非鱼链球菌的早期预警分子检测。
4 结 论
本研究建立的分子检测技术,即根据海豚链球菌16S rRNA基因中有别于无乳链球菌的特异性片段、无乳链球菌Sip基因的特异性序列设计双重PCR的引物,实现在一个反应体系中同时鉴别罗非鱼两种链球菌的目的。该检测技术具有较高的特异性、灵敏度、可重复性和稳定性,比常规诊断方法更加简便快捷,而且可检测更低含量的无乳链球菌和海豚链球菌,适用于罗非鱼链球菌的早期分子诊断和预警,将为罗非鱼链球菌病的预防和诊断提供技术支持。
