用第一性原理研究应变和Pt边缘掺杂对石墨烯氧还原能力的影响
2021-07-13杨可可刘伟伟
杨可可,刘伟伟
(杭州电子科技大学 材料与环境工程学院,杭州310018)
燃料电池作为燃烧能源的替代品,具有效率高和环保清洁等特点[1]。质子交换膜燃料电池是一种新型的燃料电池,具有环境污染小、运行噪声低及转化效率高等优点,受到了人们的广泛关注[2-4],但该电池的性能受阴极氧还原反应能力的影响较大[5]。使用铂(Pt)基材料可以有效提高阴极氧还原反应能力,但由于Pt成本较高、资源有限,制约了Pt在商业上的大规模应用[6]。近年来,为获得高效低廉的催化剂,研究人员正在探索用其他元素代替Pt基材料,或减少Pt基材料的使用。目前,通过Pt单原子催化,已可以在提高催化活性的同时,大幅减少Pt基材料的使用,使生产成本大大降低[7]。此外,通过非金属元素掺杂石墨烯进行氧还原反应催化也达到了较好的效果[8]。质子交换膜燃料电池的阴极氧还原反应主要通过双电子和四电子2种途径进行,最终产物都是水[9-17]。双电子途径中,氧气被还原为过氧化氢,会腐蚀电催化剂,影响质子交换膜燃料电池的使用寿命[18]。四电子途径可以有效避免腐蚀,比双电子途径更有优势。
石墨烯是一种新型的2维材料,具有优异的物理化学性能[19]。单层蜂窝结构的石墨烯已广泛应用于各种领域[20-22]。单原子掺杂可以调节石墨烯的电子性质和表面极性,引入带正电荷的位点可以提高定域自旋密度,这对进一步提高石墨烯作为电催化剂的催化活性具有重要作用[23]。由于石墨烯具有合成简单、能耗低和可大规模生产等特点,单原子掺杂石墨烯已发展成为一种简便有效的氧还原反应电催化剂[24]。施加应变,可以打开石墨烯的带隙,从而对石墨烯的电子结构和光学性质产生影响[25]。对于单层蜂窝状石墨烯,用N元素替换C元素,可以将无催化活性的石墨烯转变为燃料电池阴极氧还原反应的电催化材料[26-28]。用Fe, Co, Ni等过渡金属元素与N元素共掺杂石墨烯,可以进一步提高催化活性[29]。
本文采用基于密度泛函理论的第一性原理方法,系统地研究了应变和Pt原子掺杂对石墨烯氧还原反应能力的影响。
1计算方法
本文依据密度泛函理论,利用维也纳从头算模拟包(Vienna ab-initio simulation package, ASP),在自旋极化近似下进行模拟计算[30]。平面波截断能选取为400 eV,k点网格取为1×1×1。利用共轭梯度算法对原子的位置进行优化,直至总能量小于10-5eV,原子力小于0.01 eV·(Å)-1。选取xy平面为石墨烯平面,z方向为法线方向。单层平面石墨烯的晶格常数为:a0=24.65 Å,b0=22.81 Å,c0=11 Å,引入应变后的晶格常数为:a0=23.15 Å,b0=22.81 Å,c0=11 Å。为消除层间相互作用,在法线方向增加了一个厚度为11 Å的真空层。
应变ε的计算公式为
(1)
其中,ε为负数,表示压应变;ε为正数,表示拉应变。
形成能Ef的计算公式为
Ef=Etot-Ev-EPt
(2)
其中,Etot为Pt原子取代C原子边缘掺杂石墨烯体系的总能量;Ev为石墨烯有一个空位时的能量;EPt为单个Pt原子的能量。Ef为负数,表明掺杂后的结构比较稳定。
在碱性介质中,氧还原反应通过四电子反应途径进行时,可分为5个步骤:

吸附能Eads的计算公式为
Eads=Etot-Es-Ea
(3)
其中,Es为Pt原子边缘掺杂石墨烯体系的总能量;Ea为氧还原反应中间体的能量。Eads为负数时,表示有较强的吸附放热作用。
为了评价催化性能的好坏,计算了氧还原反应过程中的反应自由能。自由能ΔG的计算公式为
ΔG=ΔE+ΔEz-TΔS-QU
(4)
其中,ΔE,ΔEz分别为氧还原反应过程中生成物与反应物的能量之差和零点能之差,ΔE可通过密度泛函理论计算得到;T为热力学温度,T=298.15 K;ΔS为生成物与反应物的熵差;U和Q分别为电极电位和转移的电荷。
2结果与讨论
2.1Pt掺杂石墨烯的形成能
Pt边缘掺杂石墨烯后的结构,如图1所示。对Pt边缘掺杂石墨烯的结构进行了优化,并分别计算了去掉Pt原子形成一个空位后体系的能量和单个Pt原子的能量。单个Pt原子能量的计算中,首先计算了Pt晶体的能量,然后利用Pt晶体的能量除以Pt原子的个数,得到单个Pt原子的能量为-5.797 eV。根据式(2)计算得到图1(a)的形成能为-0.02 eV,图1(b)的形成能为-0.626 eV。因此,Pt边缘掺杂后,图1(b)的结构更加稳定,是最佳的掺杂位点。本文后续的研究中,选取图1(b)结构作为氧还原反应的基底,并对其催化性能进行研究。

(a)Structure 1

(b)Structure 2
2.2Pt边缘掺杂石墨烯对O2的吸附能

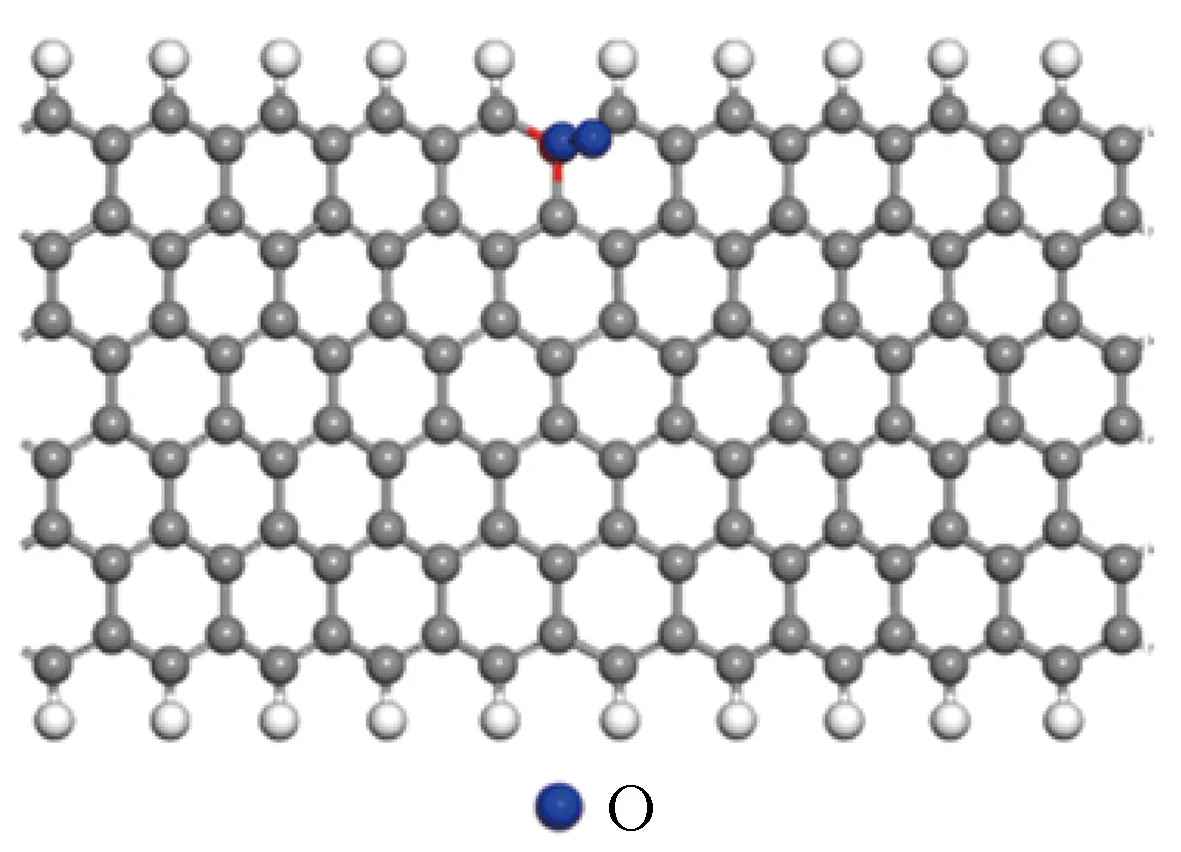
(a)Structure 1
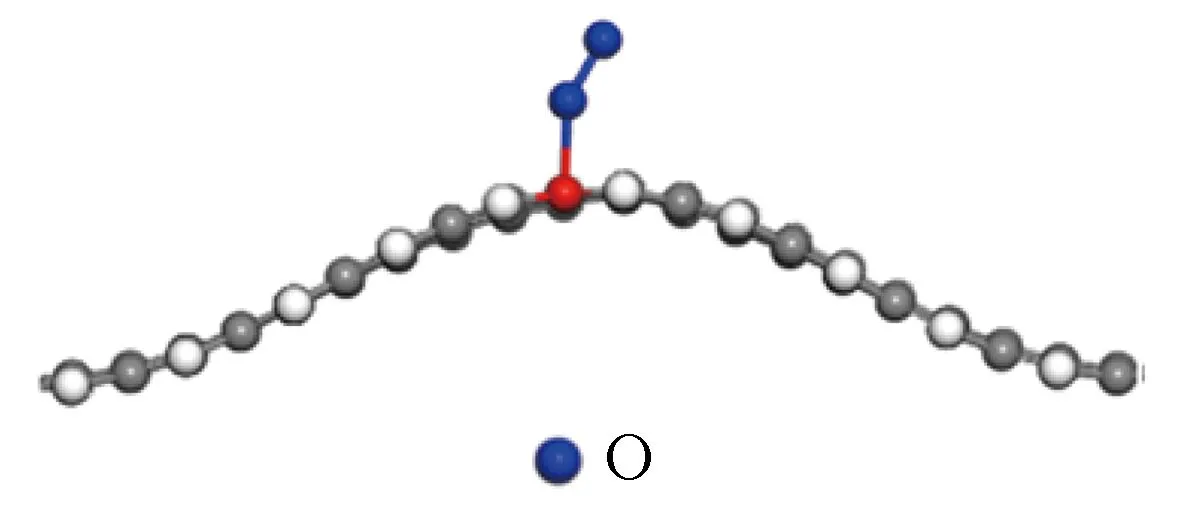
(b)Structure 2

图3不同应变下基底吸附O2的吸附能Fig.3Adsorption energies of O2 in different strains
2.3氧还原反应自由能
Pt边缘掺杂石墨烯氧还原反应过程中的自由能计算结果,如图4所示。

图4不同应变下氧还原反应的自由能Fig.4Free-energy for oxygen reductionreaction in different strains
由图4可见,对于Pt边缘掺杂石墨烯结构,当引入-6.1%的压应变时,催化性能最佳,氧还原反应的第1步放出的能量为-0.166 eV;当压应变增大到-16.2%时,优化后的晶格常数为:a0=20.66 Å,b0=22.81 Å,c0=11 Å,氧还原反应的第3步出现了吸热现象,不利于氧还原反应的进行。对于没有引入应变的Pt边缘掺杂石墨烯结构,氧还原反应的第1步放出的能量为-0.077 eV;当引入-6.1%的压应变时,第1步放热更多,催化效果更好。对于纯净的石墨烯结构,氧还原反应的第4步出现了吸热现象,不利于氧还原反应的进行。氧还原反应的各步能量变化,如表1所列。
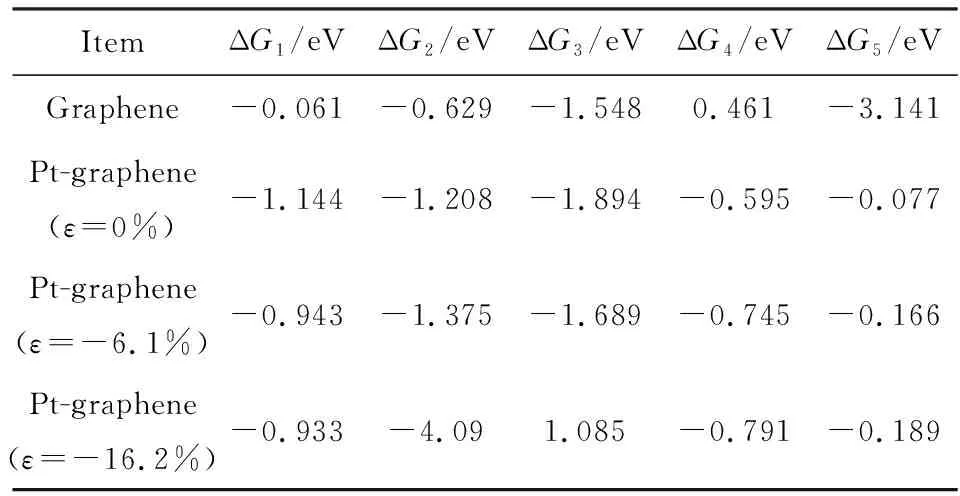
表1氧还原反应各步的能量变化Tab.1Energy change in oxygen reduction reaction
2.4Pt边缘掺杂石墨烯的电子态密度
通过对电子态密度的计算,分析引入应变对Pt边缘掺杂石墨烯电子结构的影响。图5为不同应变下Pt边缘掺杂石墨烯的电子态密度。

图5不同应变下Pt边缘掺杂石墨烯的电子态密度Fig.5Density of states of Pt-doped graphene in different strains
由图5可见,随着压应变的引入,费密面附近的电子态密度峰值向左侧移动,费密面左侧为价带,右侧为导带。引入应变促进了Pt边缘掺杂石墨烯作为燃料电池阴极氧还原反应电催化剂的电子转移,有利于催化反应的进行。
3结论
本文利用基于密度泛函理论的第一性原理方法,研究了应变和Pt边缘掺杂对石墨烯氧还原反应的影响。结果表明,引入应变后基底吸附O2之后的结构是稳定的;当引入-6.1%的压应变时,Pt 边缘掺杂石墨烯的催化性能最佳。电子态密度的计算结果表明,随着压应变的引入,费密面附近的电子态密度峰值向价带移动,促进了Pt边缘掺杂石墨烯作为燃料电池阴极氧还原反应电催化剂的电子转移,有利于催化反应的进行。
