黏多糖贮积症Ⅱ型的诊断及治疗进展
2021-07-09巩纯秀李晓侨
巩纯秀 李晓侨
黏多糖贮积症Ⅱ型(mucopolysaccharidosis type Ⅱ,MPSⅡ,MIM #309900)于1917年由加拿大医生Charles Hunter首次报道,亦称为亨特综合征(Hunter 综合征)。该疾病是一种溶酶体贮积症,因编码艾杜糖醛酸-2-硫酸酯酶(iduronate-2-sulfatase,IDS)的基因——IDS基因(MIM*300823)发生变异,致溶酶体内IDS酶活性缺乏或显著降低,从而使该酶催化硫酸皮肤素的2-硫酸盐基团(DS)和硫酸乙酰肝素(HS)的水解能力下降,导致这两种糖胺聚糖(曾称为黏多糖)不能降解而堆积在溶酶体中,导致溶酶体肿胀、细胞破坏及脏器功能损害,并引起一系列临床表现[1]。
有调查表明,男性发生率为1∶(100000~170000)[2]。近年来,国内外有关该疾病的治疗取得了很多新进展。2006年,英国制药公司夏尔(Shire)研制的药物Elaprase获美国食品药品监督管理局(Food and Drug Administration, FDA) 批准,成为全球获批的首个MPSⅡ酶替代治疗药物。在亚洲也逐渐开展了酶替代疗法的临床试验[3]。近年来,随着基因治疗的开展,基因编辑产品亦成为该疾病的研究热点,并逐渐应用于临床。本文就总结该疾病的临床表现,实验室检查,重点介绍目前该疾病的治疗进展,以供临床医生参考。
一、临床表现及分型
MPSⅡ患儿在大多数器官系统中可能受到不同程度的影响,疾病表现存在很高的异质性。根据中枢神经系统是否受累,可将患儿分为早期进展型和缓慢进展型,前者主要表现为进行性认知恶化,加上进行性气道和心脏疾病,通常会导致患儿在<20岁死亡。缓慢进展型患儿中枢神经系统影响较小,对其他脏器的影响也较轻,通常智力正常,可以活到成年[2]。
MPSⅡ患儿GAG的堆积几乎可以发生在所有器官中,但是对某些特定脏器影响尤为明显。
1.面部改变:新生儿期外观可正常,面部特征的粗陋化在早期进展型中多在18个月~4岁出现,而缓慢进展型可晚几年出现。主要表现为舌大、眶上嵴突出、鼻大、鼻梁宽以及脸颊大而圆和嘴唇厚。部分患儿的上臂背侧和外侧面可出现象牙色的皮肤病变。
2.生长:患儿出生时表现正常,在出生后的头几年,大多数患儿的身高都在第50百分位以上,有的甚至超过了第97百分位。然而,生长速率随着年龄的增长而降低。通常到8岁时,身高可低于第3百分位,几乎所有的儿童在青春期前都表现出生长迟缓,且都伴有大头畸形。身材矮小的原因不明,可能与骨生长板紊乱有关。虽然在缓慢进展型和早期进展型中并没有观察到身高之间的差异,但生长的监测仍有助于帮助评估疾病进展和治疗效果。
3.眼睛:偶有MPSⅠ常见的角膜浑浊。但是,裂隙灯检查可能会发现散在的角膜病变,一般不影响视力。大约20%的个体存在视神经盘水肿(papilledema),但不伴颅内压增高。大约11%的患者可有视神经萎缩。视网膜病变常见于早期进展型,视网膜电图(ERG)异常可以揭示视网膜功能障碍。其他眼部异常包括双侧葡萄膜积液、周围色素上皮改变和放射状中央窝旁皱褶。
4.口腔、耳鼻喉:常见舌骨增大,腺样体和扁桃体肥大以及颞下颌关节强直,导致张口困难。咽喉中GAG沉积会导致声音嘶哑。患儿牙齿形状不规则,牙龈组织增生及牙源性囊肿。患儿可出现传导性和感觉神经性听力损伤,并伴有耳部反复感染。蛛网膜增生引起的耳蜗神经受压以及螺旋神经节细胞数量减少和毛细胞变性可导致神经性耳聋。
5.关节及骨骼:关节挛缩是最早出现的症状,尤其指骨关节挛缩。挛缩导致关节活动显著丧失。骨骼异常并非是MPSⅡ的特异表型,这种在影像学上表现的“多发性骨发育不良”在所有的MPS患者中均可出现,主要表现为:大部分长骨,尤其是肋骨在胸骨端的广泛增宽,脊柱端相对偏细,形如“飘带”状。掌指骨短粗,远端宽,近端尖呈三角形,远节指骨呈爪形。楔形蝶鞍、颅骨呈舟状。许多区域的骨骺骨化中心不规则。椎体可见切迹。髋关节发育不良,如果不治疗,可发展为早发性关节炎,从而导致严重的残疾。
6.呼吸系统:随着GAG在舌、口咽软组织和气管中积累,气道逐渐变窄,分泌物增加,最终导致气道阻塞,频发上呼吸道感染。另外,胸壁僵硬,肝脏、脾脏肿大,从而减少胸腔容积。若出现睡眠呼吸暂停,需正压辅助通气,最终可能需气管切开术。
7.心血管疾病:心脏异常是主要的临床表现,同时也是死亡的主要原因。据报道,82%的患儿有心血管异常,其中62%与瓣膜疾病有关。此外,心肌病、高血压、心律失常和外周血管疾病也有少数报道(<10%)。
8.胃肠道:大多数受累患儿会出现肝大和(或)脾大,脐或腹股沟疝也常出现。在早期进展型MPSⅡ患儿中,慢性腹泻为常见主诉。
9.神经系统:患儿在出生时表现正常,早期的发育也可能在正常范围内。里程碑发育延迟通常是大脑受累的第一个表现。随着认知功能的损害逐渐加重,患儿可出现睡眠障碍、多动、大运动落后、有癫痫样发作、持续的咀嚼动作,不能控制排便。通常患儿在6~8岁出现发育倒退。
腕管综合征(CTS)是MPS Ⅱ常被忽视的并发症,神经传导检查可提示其不正常,通常术后手功能可改善。另外,椎管狭窄,尤其是颈部脊髓受压必须注意监测。
二、确诊方法
1.IDS酶活性测定:除了成熟的红细胞外,IDS蛋白存在于所有细胞中,可以在不同的细胞或体液中评估其酶活性,在进行酶学检查时需对至少一个甚至多个硫酸酯酶与IDS的活性一起检测,以排除多种硫酸酯酶缺乏症(MIM#272200)。大多数MPSⅡ患儿没有残留的IDS活性,轻型患儿可能残存0.2%~2.4%的酶活性[4]。
2.IDS基因分子遗传检测:由于IDS基因是目前报道MPSⅡ的唯一致病基因,因此通过一代测序方法可直接检测IDS基因变异。因其存在假基因,可以用Lualdi等[5]提出的基于PCR的简单重组子分析法检测。阵列CGH(比较基因组杂交)分析方法可检测基因的大缺失和(或)重复。随着二代测序(NGS)技术的发展和普及,近几年可直接用二代测序方法检测,尤其对非典型临床特征的患儿。
三、鉴别诊断
MPSⅡ症状体征最广泛,包含了所有其他类型黏多糖贮积症和溶酶体储存障碍的表现,如黏脂病Ⅱα/β、Ⅲα/β和Ⅲγ、甘露糖病、岩藻糖病和多种硫酸酯酶缺乏症,因此需要通过酶学检查或基因检测确诊。
还需与伴有大头畸形和(或)器官肿大并表现为发育延迟的其他非溶酶体疾病相鉴别。尿GAG定性分析可鉴别出不同类型的MPS,而不仅仅是区分MPSⅠ和MPSⅡ。确诊则要通过对酶活性检测及遗传分析[2]。
四、治疗和管理
1.对症治疗:由于目前常见的治疗干预措施无法到达患者脑室,因此与神经受累有关的症状仍无法治愈,只能采取对症治疗。脑积水和脊髓压迫可进行减压手术,癫痫可加用抗惊厥药物,肌环切开术改善呼吸,眼科手术改善视网膜病变[6]。因为镇静或麻醉对患者非常危险,所以对所有需要手术干预的患儿治疗前都要进行仔细评估。
骨骼功能障碍主要表现为发育不良和活动受限,后者用物理治疗。在有髋关节畸形的情况下,才进行骨科手术、腹股沟和脐疝手术治疗[7]。
2.酶替代疗法(ERT):针对MPSⅡ研制出两种不同的重组酶,分别为聚硫酶和聚硫酶β。这两种酶在临床前期研究中都有相似的理化性质,在降低GAG水平方面具有相似的器官分布和功效,而聚硫酶β则表现出更高的比酶活性、更快的细胞摄取率以及形成更低的抗药物抗体,并且研究表明获得较好的结果[8~11]。在安全性方面,ERT耐受性较好,不良反应在轻度至中度[12]。2017年美国医学遗传学和基因组学学院(ACMG)基金会委托专家发表了关于MPSⅡERT的系统证据综述,结果表明,在MPSⅡ患儿中,每周静脉注射替代酶会降低uGAG水平和肝脏/脾脏体积,而对于其他结果证据并不太明确[13]。
因为骨、软骨和心脏瓣膜等组织由于其血管化程度低,因此治疗酶的生物利用度也低。而血-脑脊液屏障的存在对于CNS症状的ERT也存在困难[14]。
重组酶的免疫反应性也会减弱治疗效果。超过50%的ERT患者出现了抗硫酸酶抗体IgG[3]。注射重组酶在输注完成后可能会被机体立即清除,需要频繁输注[14]。所以经常住院持续输液治疗和治疗费用高,限制了治疗的可及性。对于MPSⅡ重型患儿,ERT的治疗仍有争议。但目前科学界的立场仍是在没有找到更有效治疗方法前所有的患者都建议进行ERT[15]。
为了克服 ERT方法的局限性,研究者对传统ERT治疗进行了一些改进,包括改变给药途径及引入修饰的融合蛋白替代宿主进行酶的生产。 为了使ERT能治疗中枢神经系统症状,研究者还进行了对脑室内(ICV)和鞘内(IT)注射治疗。在MPSⅡ小鼠模型中,脑室内给药和鞘内给药均获得较好的阳性结果[16,17]。在Ⅰ/Ⅱ期的临床试验(包括安装鞘内给药装置)显示出良好的效果,目前仍在进行的Ⅱ/Ⅲ期试验将于2022年结束[1]。
用分子木马(TH)修饰治疗性重组酶形成融合蛋白可跨越血-脑脊液屏障(BBB)。分子TH是一种内源性分子(一般是肽链或单克隆抗体),可结合位于大脑毛细血管内皮细胞腔内的内源性受体,通过受体介导的转运方式穿过BBB。 目前主要用于这一目的的受体是胰岛素受体(IR)和转铁蛋白受体(TfR),它们可以作为特异性单克隆抗体受体在BBB上转运重组蛋白药物[18]。该蛋白在恒河猴试验中已证明其安全性,且在老鼠研究中获得较好结果[19,20]。目前,为了降低ERT的成本而生产出改善稳定性、药代动力学及药效学特性的蛋白质仍在研究中。
3.造血干细胞移植(HSCT):HSCT包括将血液干细胞从相容的健康供体移植给患者,理论上,移植细胞和(或)其后代细胞可以在局部产生永久功能性治疗的酶,目前通常使用骨髓外周血或脐带血作为血液干细胞来源。移植后血管周围间隙和脑实质的小胶质细胞中存在供体细胞,提示HSCT具有治疗MPSⅡ神经症状的潜力[21]。在1986年首次用 HSCT 治疗1例7岁MPSⅡ患者,移植后3.5年因心血管并发症死亡[13]。因此HSCT一直受阻。一项研究收集了146例HSCT治疗的MPSⅡ患儿与51例ERT和15例未经治疗的病例进行比较,该研究认为,HSCT对于MPSⅡ患儿似乎比ERT更有效,HSCT可以作为该疾病的治疗选择[22]。中国儿童移植学组(CCTG)对收治的34例MPS患儿(包括12例Ⅱ型)的移植资料进行分析,结论得出同种异体HSCT可以挽救MPS患儿的生命,提高生活质量[23]。因此,2017年,中华医学会儿科学分会血液学组发表了HSCT治疗MPS的专家共识,并提出该治疗方案目前仍是我国提供给患儿的一种治疗方式[24]。但进一步改善移植方案,降低其发生率或病死率的风险,仍是目前需要解决的问题。
4.基因治疗:MPSⅡ以及其他类型MPS和大多数LSD,由于都是单基因疾病,使他们成为候选基因治疗的潜在疾病,即经过基因修饰后可以由一个储存器官的细胞产生和释放缺陷的酶,然后被其他细胞和器官吸收达到治疗的目的。此外,诱导基因表达水平不用特别充足就可以纠正临床表现,通常仅需要正常酶5%~15%的水平就可以维持正常[12]。
基因治疗方法可分为体内和体外两种。前者指将携带治疗基因的载体直接输注到患者体内,主要使用反转录病毒、慢病毒、腺病毒和基于腺相关病毒(AAV)的载体以及非病毒载体系统来传递。后者是从受体患者中提取细胞,通常是造血干细胞或外周血细胞,用治疗基因进行体外转导后再注入回患者体内,主要使用反转录病毒和慢病毒载体[1]。 此外,细胞治疗和纳米载体、减少底物基质治疗、药理学伴侣治疗等治疗方法目前都仍在研究中(表1)。
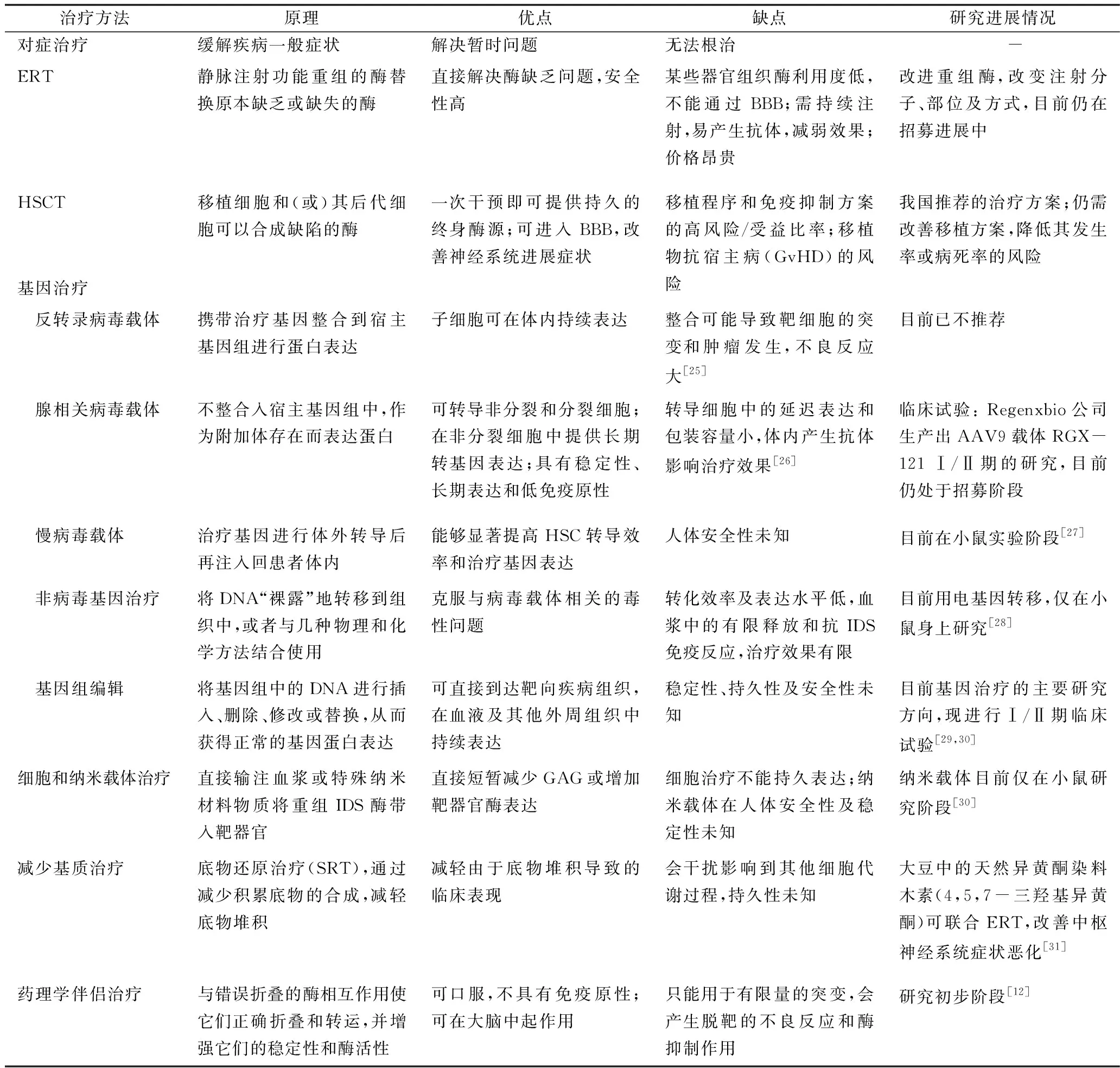
表1 MPSⅡ的治疗方法
五、展 望
对于MPSⅡ的发病机制以及临床和实验室诊断方面,已经研究出很多治疗该疾病的方法。但是,仍有一些问题需要解决,多年来,该疾病的发生机制一直仅考虑到未降解的GAG在不同器官中积累,现在已知该疾病的发生机制可能更为复杂,且存在更多未预料到的方面,对这些致病机制的深入研究有望鉴定出其他更具体的治疗靶标。而且鉴于在同一家系中出现不同程度的中枢神经系统的受累患者,因此,表观遗传因素可能也需要进行研究。
从治疗的角度来看,在开始ERT方案后14年,需要重新评估和考虑不同的剂量和(或)给药频率。此外,重新定义ERT患者入组或停药指南也很重要。而对于发展中国家,随着HSCP治疗方案的优化,HSCP也可以作为该疾病的首选治疗方式之一。当然,随着基因治疗研究的不断深入,患者有望在未来能一次性改善糖胺聚糖代谢能力,达到彻底治愈。
