非小细胞肺癌差异表达基因的生物信息学分析
2021-07-09郑浩然蒋爱民阮之平
郑浩然,蒋爱民,傅 潇,田 涛,梁 璇,阮之平,姚 煜
(西安交通大学第一附属医院肿瘤内科,陕西西安 710061)
肺癌已经成为全世界癌症死亡的主要原因,其发病率和死亡率近年来显著升高[1]。一系列的研究表明,吸烟、空气污染、职业暴露等因素均与肺癌发生有关[2]。所有肺癌患者中,非小细胞肺癌(non-small cell lung cancer, NSCLC)约占肺癌病理分型的85%[3]。早期NSCLC患者接受手术治疗后预后尚可[4]。近年来,尽管NSCLC的早期诊断和治疗取得了较大进展,但其预后仍不容乐观。因此,寻找能够准确预测患者预后的生物标志物至关重要。随着科技的发展,大量基因芯片数据库的建立为研究肺癌差异表达基因(differently expressed genes, DEGs)提供了重要基础。本研究在GEO数据库中选取了GSE19804和GSE33532两个肺癌基因表达谱芯片作为研究数据集,筛选出DEGs并探讨其在NSCLC发生发展过程中的功能及其与患者预后的关系,从而为NSCLC的靶向治疗提供新策略。
1 资料与方法
1.1 芯片数据的选择本研究使用的是来自美国国立生物技术信息中心的GEO数据库(https://www.ncbi.nlm.nih.gov/geo)中的基因芯片数据,系列号分别是GSE19804和GSE33532。其中GSE19804数据集由LU等[5]于2011年发表,收集了60例NSCLC样本和60例正常肺组织样本。GSE33532数据集由MEISTER等于2014年发表,收集了80例NSCLC样本和20例正常肺组织样本。
1.2 方法
1.2.1NSCLC与正常肺组织差异基因的筛选 采用GEO2R(https://www.ncbi.nlm.nih.gov/geo/geo2r/)对NSCLC组织与正常肺组织之间的DEGs进行筛选。利用默认的Benjamini和Hochberg错误发现率方法,调整P值来降低假阳性率。以调整后P<0.05、|log2FC|≥2作为截断标准,运用FunRich3.1.3对两个数据集中的DEGs取交集,最终筛选出共同的DEGs。
1.2.2差异基因的富集分析 采用生物信息注释数据库DAVID(https://david.ncifcrf.gov/)对DEGs进行基因本体(gene ontology, GO)分析及京都基因和基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)分析,并对共表达DEGs的功能分析(细胞组分、分子功能、生物学过程以及信号通路)进行可视化,以P<0.05及错误发现率(false discovery rate, FDR)<0.05为显著性基因富集[6]。
1.2.3PPI网络的构建及枢纽基因的筛选 采用STRING(search tool for the retrieval of interacting genes/proteins)10.5(https://string-db.org/)在线工具对DEGs进行蛋白质-蛋白质相互作用网络(protein-protein interaction, PPI)分析。以置信度得分≥0.4且最大相互作用数=0作为界值对DEGs进行PPI分析。随后,运用Cytoscape3.8.0(http://www.cytoscape.org/)[7]中的cytohubba插件对PPI网络进行分析,筛选出与周围基因有高度连通性(度值degree)的前20个基因作为枢纽基因。cytohubba通过几种拓扑算法预测和探索给定网络中重要节点和子网之间的相互关系。在网络拓扑理论中,度(connect degree, k)被定义为某节点与网络中其他节点之间相互连接的数目,即相邻蛋白质的数量。
1.2.4枢纽基因的生存分析 运用Kaplan-Meier plotter数据库(http://www.kmplot.com/analysis/)对筛选出的20个枢纽基因进行预后分析,选择与NSCLC患者总生存期(overall survival, OS)呈相关性(P<0.05)的基因,并通过Kaplan-Meier法绘制生存曲线。
1.2.5通过多个外部数据库对枢纽基因表达水平及其与预后关系进一步验证
1.2.5.1GEPIA数据库 GEPIA基因表达谱数据动态分析(http://gepia.cancer-pku.cn/)是由北京大学张泽民教授团队开发的用于癌症和正常组织基因表达以及交互式分析的网络服务器,提供交互式和自定义功能,包括差异基因表达分析、生存分析等[8]。本研究运用GEPIA分析枢纽基因在NSCLC和正常组织中的表达差异,同时进行生存分析。
1.2.5.2HPA数据库 运用HPA数据库(https://www.proteinatlas.org/)初步验证枢纽基因在NSCLC和正常组织中的蛋白质表达情况。
1.2.5.3UALCAN数据库 UALCAN(http://ualcan.path.uab.edu/)是基于TCGA数据库和CPTAC数据库进行癌症数据在线分析和挖掘的网站。本研究通过UALCAN对枢纽基因及其蛋白在NSCLC和正常组织中的表达情况再次进行验证。
1.2.6统计学分析 在线数据库分析均应用系统默认的统计学方法。通过Kaplan-Meier法进行生存分析,采用对数秩和检验,以P<0.05差异有统计学意义。
2 结 果
2.1 差异基因的筛选从GSE19804和GSE33532两个数据集中分别筛选出264和795个DEGs,分别包括212、547个上调基因和52、248个下调基因。图1A和图1B为两个数据集差异基因的火山图,横坐标表示-Log10(P.value),纵坐标表示log2(fold-change)(NSCLCvs.正常样本),绿色表示下调的DEGs,红色表示上调的DEGs。运用FunRich3.1.3软件对两个数据集中的DEGs取交集并绘制韦恩图(图1C),最终筛选出共同的DEGs共159个。

图1 差异基因的筛选
2.2 差异基因的富集分析
2.2.1GO分析 对159个共同DEGs进行GO分析,发现它们在细胞组分上(图2A)主要在胶原蛋白三聚体、细胞膜边界的囊泡腔、细胞外基质蛋白等处富集;在分子功能上(图2B)主要在氧气运输、CXCR趋化因子受体整合、氧气整合、细胞外基质整合等方面富集;在生物学过程中(图2C)主要在细胞外基质组装、细胞外结构组装、血管形态发育等方面富集。
2.2.2KEGG分析 KEGG分析(图2D)显示共有6条通路呈显著相关,包括疟疾、非洲锥虫病、细胞外基质相互作用、过氧化物酶体增殖物激活受体(PPAR)信号通路、蛋白质消化吸收及病灶黏附。
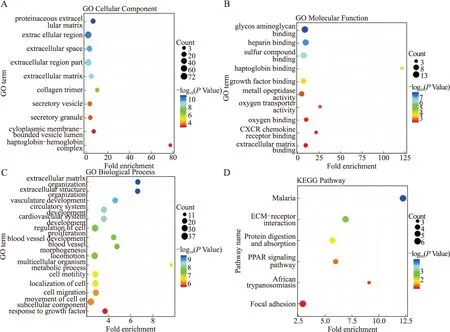
图2 差异基因的GO和KEGG富集分析
2.3 PPI网络的构建及枢纽基因的筛选登录STRING网站(https://string-db.org/),选择Multiple Proteins,输入经初步筛选出的159个共同DEGs,物种选择Homo sapiens,初步构建PPI网络图。为发现NSCLC发展过程中的潜在调控基因,将STRING中的159个DEGs导入Cytoscape3.8.0以构建PPI网络(图3)。运用cytohubba插件对PPI网络进行运算,按degree方法选择前20个基因(图4,表1),并将这20个基因作为枢纽基因。

表1 前20个枢纽基因(按degree排序)

图3 PPI网络
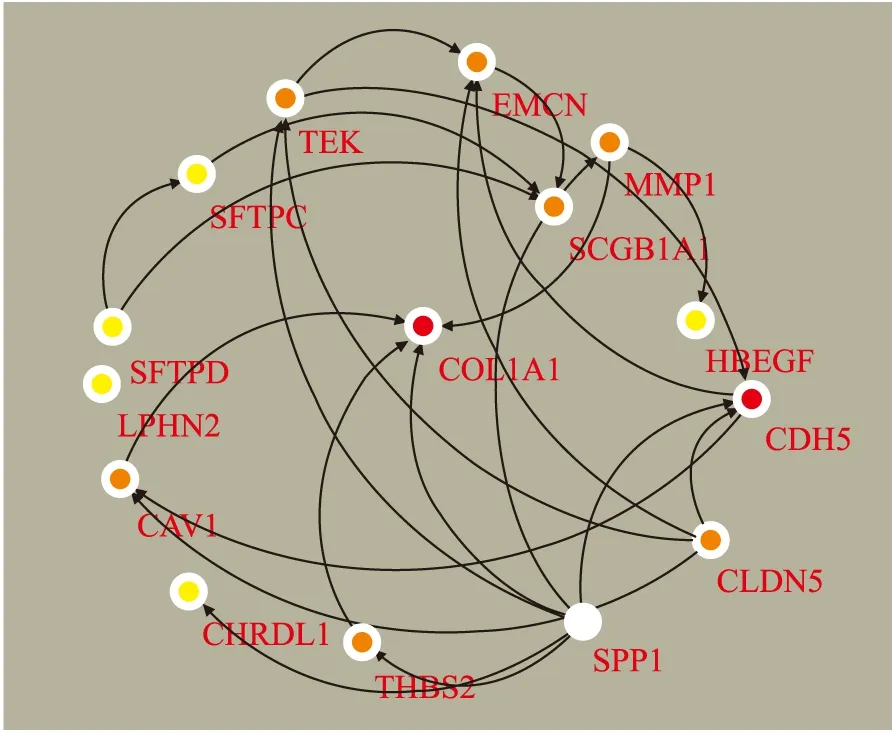
图4 前20个枢纽基因
2.4 枢纽基因的生存分析运用Kaplan-Meier plotter对筛选出的20个枢纽基因进行生存分析,结果表明,20个枢纽基因中有15个基因与患者的OS呈相关性(P<0.05)。其中COL1A1和SPP1(HsT2645)基因高表达与患者更短的OS呈相关性(图5),表明这两个基因可能在NSCLC的发生发展中发挥潜在的促进作用;其余的13个枢纽基因高表达则是患者OS的保护性因素。
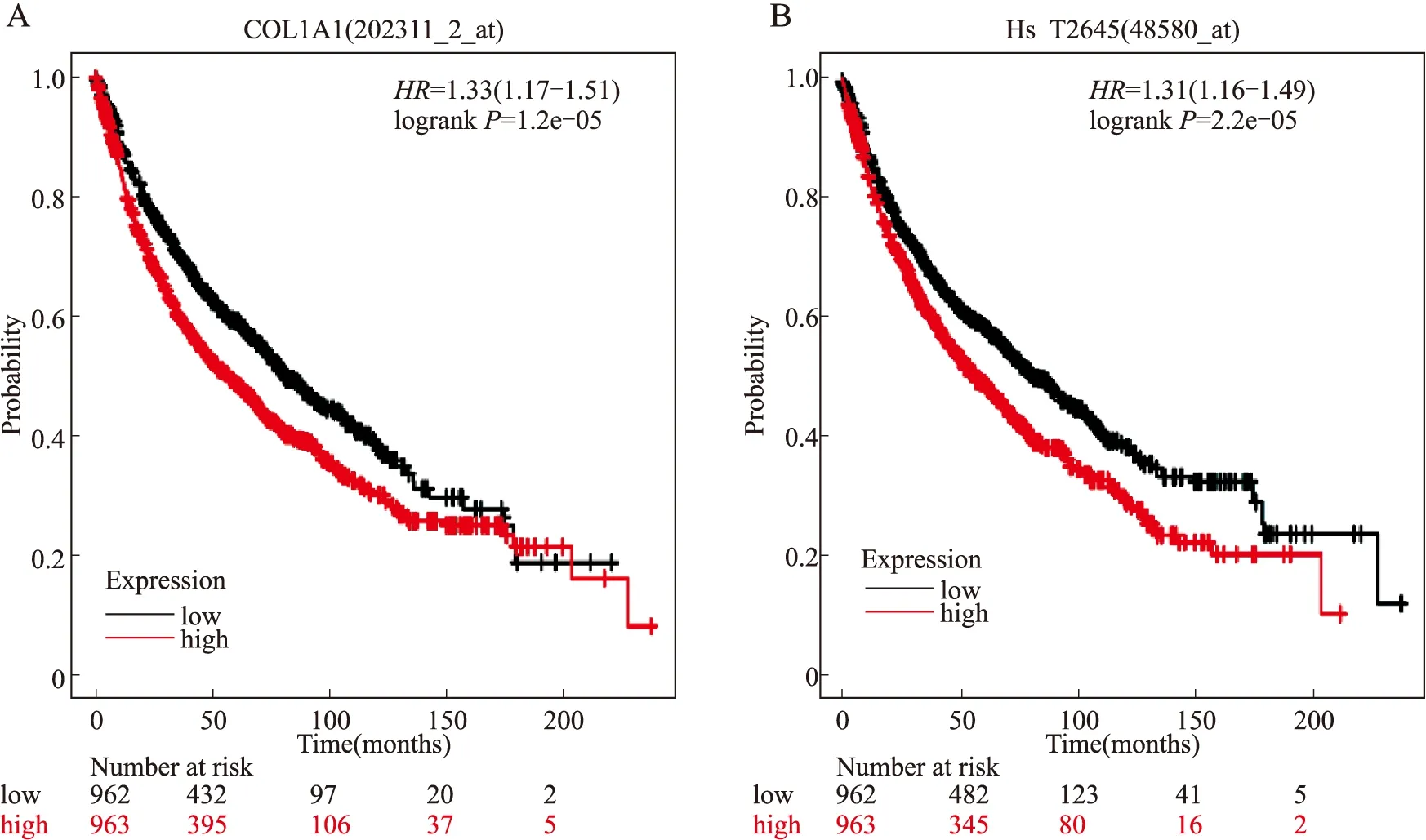
图5 枢纽基因的生存曲线(Kaplan-Meier plotter)
2.5 通过多个外部数据库对枢纽基因表达水平及其与预后关系的进一步验证运用GEPIA数据库分析NSCLC和正常肺组织中COL1A1和SPP1基因的表达水平(图6)。结果显示,COL1A1和SPP1在NSCLC组织中的表达水平是上调的(P<0.05)。生存分析(图7)显示,COL1A1和SPP1基因高表达与患者更短的OS呈相关性(两组HR=1.3,P<0.05)。进一步支持Kaplan-Meier plotter数据库分析结果。

图6 枢纽基因在NSCLC和正常组织的表达水平(GEPIA)
运用HPA数据库初步验证COL1A1和SPP1基因在NSCLC和正常组织中的蛋白质表达情况,发现COL1A1蛋白在多种癌症组织及正常组织中均有表达,特异性较低,而且已被证实与肺癌不良预后相关(P<0.001)。为此,本研究只展示了SPP1蛋白在NSCLC和正常组织中的免疫组化结果(图8)。

图8 免疫组化:SPP1蛋白在NSCLC和正常组织中的表达(HPA)
通过UALCAN分别分析了TCGA数据库中515例肺腺癌组织与59例正常组织(图9A)、503例肺鳞癌组织与52例正常组织(图9B)中SPP1基因的表达情况。结果显示,SPP1基因在NSCLC组织中的表达水平是上调的(两组P<0.01)。分析CPTAC数据库中111例肺腺癌组织与111例正常组织中SPP1蛋白的表达情况(图9C),结果显示,SPP1蛋白在NSCLC组织中的表达高于正常组织(P<0.01)。亚组分析显示,淋巴结N3组SPP1基因表达高于N1、N2组(P<0.05),说明SPP1可能通过影响淋巴结转移导致患者预后不良。

图9 SPP1基因及蛋白在NSCLC和正常组织的表达水平(UALCAN)
3 讨 论
目前,肺癌已经成为全世界范围内癌症相关死亡的首要原因。但是,由于多数NSCLC患者确诊时已经处于晚期,无手术机会,5年生存率仅为16%[9-10]。肺癌组织复杂的生物学行为涉及多种基因及相关通路[11],目前对于其发生发展机制尚不十分清楚。随着大数据时代的到来,基因数据库的共享使得从基因水平揭示肺癌的发生发展成为当下研究者研究的热点。本研究旨在运用生物信息学分析的方法筛选出NSCLC与正常肺组织之间的差异基因,进而探索与NSCLC预后相关的生物标志物,从而为NSCLC的诊疗提供新思路。
本研究对GSE19804和GSE33532两个基因芯片进行挖掘,共筛选出NSCLC与正常肺组织之间的159个DEGs。通过STRING和Cytoscape3.8.0构建DEGs的PPI网络并进行计算,最终确定20个枢纽基因。对20个枢纽基因运用Kaplan-Meier plotter进行生存分析发现,COL1A1和SPP1(HsT2645)两个基因的高表达与患者更短的OS显著相关。经GEPIA分析,二者在NSCLC与正常肺组织之间的表达差异有统计学意义,生存分析结果与Kaplan-Meier plotter一致。但HPA数据库显示,COL1A1蛋白在癌组织与正常组织中表达缺乏特异性,与基因表达水平不一致,临床价值有限。为此,本研究最后通过UALCAN仅验证了SPP1基因及其蛋白的表达情况。结果显示,SPP1基因及其蛋白在癌组织的表达高于正常组织。
SPP1(分泌磷蛋白1)主要位于细胞外基质,主要参与细胞基质附着、骨化、抗细胞凋亡、细胞附着、免疫细胞趋化等生物学过程。据报道,SPP1可促进肿瘤细胞存活,调节肿瘤相关的血管生成和炎症反应[12],与肺癌、卵巢癌、胶质母细胞瘤、结肠癌的转移和不良预后密切相关[13-17]。ZHANG等[18]的研究结果表明,SPP1通过对PD-L1的上调介导巨噬细胞极化,并促进肺腺癌的免疫逃逸。以上研究表明,SPP1可能作为癌基因参与肿瘤的发生发展,本研究结果与上述研究保持一致。通过GO与KEGG分析,差异基因主要在细胞外基质方面富集,这一结果更加支持了本研究结论的可靠性。生存分析显示,SPP1基因高表达与NSCLC患者总生存(OS)存在明显的相关性,高表达组OS均低于低表达组。因此,SPP1基因在NSCLC中表达水平上调,高表达患者的总生存期更短。UALCAN亚组分析显示,淋巴结N3组SPP1表达水平高于N1、N2组,说明SPP1可能通过影响淋巴结转移导致NSCLC患者预后不良。
综上,SPP1基因可能成为评价NSCLC患者预后的生物学标志物,为NSCLC的诊断及治疗提供了新思路。
