云南白药胶囊微生物限度检查方法学研究
2021-07-08张秀花刘艳平
张秀花,刘艳平
(菏泽市食品药品检验检测研究院,山东 菏泽 274032)
云南白药胶囊收载于《中华人民共和国药典》(以下简称:《中国药典》)2020年版一部[1],国家绝密配方,一类中药保护品种,具有化瘀止血,活血止痛、解毒消肿的功效,临床应用非常广泛[2-6],目前尚无微生物限度检查方法的报道。本试验在查阅近年来中和法非无菌制剂微生物限度检查方法研究的基础上[7-12],参照企业提供的相关资料,采用3批样品进行微生物限度检查方法适用性试验,建立了适用于云南白药胶囊微生物限度检查的中和法。
1 仪器与材料
1.1 样品
云南白药胶囊,云南白药集团股份有限公司,批号ZLB1907,ZDA2012,ZHA1904。
1.2 培养基
胰酪大豆胨琼脂培养基(TSA),批号200709;胰酪大豆胨液体培养基(TSB),批号200224;沙氏葡萄糖琼脂培养基(SDA),批号200316;沙氏葡萄糖液体培养基(SDB),批号200406;肠道菌增菌液体培养基(EE),批号180222;均由北京陆桥技术股份有限公司提供,培养基适用性检查结果符合《中国药典》2020年版四部规定。
1.3 菌种
金黄色葡萄球菌(Staphylococcus aureus)[CMCC(B)26 003]、铜绿假单胞菌(Pseudomonas aeruginosa)[CMCC(B)10 104]、枯草芽孢杆菌(Bacillus subtilis)[CMCC(B)63 501]、黑曲霉(Aspergillus niger)[CMCC(F)98 003]、白色念珠菌(Candida albicans)[CMCC(F)98 001]、大肠埃希菌(Escherichia coli)[CMCC(B)44 102]、乙型副伤寒沙门菌(Salmonella paratyphi B)[CMCC(B)50 094],均由中国食品药品检定研究院提供,经鉴定合格,试验所用菌种均为第三代。
1.4 仪器
电子天平(JE1201,上海浦春计量仪器有限公司)、脉动真空灭菌器(XG1.U,山东新华医疗器械股份有限公司)、生物安全柜(BSC-1300ⅡA2,苏州安泰空气技术有限公司)、细菌培养箱(KB240,德国BINDER)、霉菌培养箱(MJ-250BSH-Ⅲ,上海新苗医疗器械制造有限公司)。
1.5 试剂
中和剂:聚山梨酯80(国药集团化学试剂有限公司,批号:20190821);稀释液:pH 7.0 无菌氯化钠-蛋白胨缓冲液(北京陆桥技术股份有限公司,批号:190206)。
2 方法与结果
2.1 菌液制备
将金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌、大肠埃希菌、乙型副伤寒沙门菌分别接种于TSB中,30~35 ℃培养 18~24 h;白色念珠菌接种于SDB中,20~25 ℃培养 2~3 d;黑曲霉接种于SDA中 20~25 ℃,培养 5~7 d。
取上述新鲜培养物用 pH 7.0 无菌氯化钠-蛋白胨缓冲液洗脱、稀释,制成5000~10 000 cfu/ml菌悬液。取黑曲霉的新鲜培养物加入 3~5 ml含 0.05 %(v/v)聚山梨酯 80 的 pH 7.0 无菌氯化钠-蛋白胨缓冲液,将孢子洗脱。吸出孢子悬液至无菌试管内,用 含 0.05 %(v/v)聚山梨酯 80的 pH 7.0 无菌氯化钠-蛋白胨缓冲液稀释,制成5000~10000 cfu/ml菌悬液。
2.2 样品前处理
取样品3批,每批4个独立包装,打开外包装,将内包装置于生物安全柜内。开启生物安全柜及高效过滤器,当洁净度符合要求时[13],以无菌操作的方式,取出胶囊及保险子,混匀,备用。
2.3 供试液制备
2.3.1 常规法 取“2.2”项下供试品10 g(至少含1粒保险子),加TSB 100 ml,待囊壳、内容物及保险子全部溶解后,混匀,制成1:10供试液。取1:10供试液5 ml至5 ml TSB中,制成1:20供试液,依法制成1:50供试液。
2.3.2 中和法 取“2.2”项下供试品10 g(至少含1粒保险子),加含5 %聚山梨酯80的TSB 100 ml,待囊壳、内容物及保险子全部溶解后,混匀,制成1:10供试液。取1:10供试液5 ml至含5 %聚山梨酯80的5 ml TSB中,制成1:20的供试液,依法制成1:50供试液。
2.4 微生物计数法
2.4.1 常规法 取“2.1”项下菌液0.1 ml,至9.9 ml“2.3.1”项下1:10供试液中,混匀,作为试验组;取TSB 0.1 ml,至9.9 ml“2.3.1”项下1:10供试液中,作为供试品对照组;取“2.1”项下菌液0.1 ml,至9.9 ml TSB中,作为菌液对照组;以上每组各取1 ml注入平皿中,照《中国药典》2020年版四部通则1105[14],加入15~20 ml对应的TSA或SDA培养基,每种菌液平行制备2个平皿,测定菌液中的菌落数,计算各试验菌回收比值。
2.4.2 稀释法 取“2.1”项下菌液0.1 ml,至9.9 ml“2.3.1”项下1:20供试液中,混匀,作为试验组;取“2.1”项下菌液0.1 ml,至9.9 ml“2.3.1”项的1:50供试液中,混匀,作为试验组;取“2.1”项下菌液0.1 ml,至9.9 ml TSB中,作为菌液对照组,依法操作,计算各试验菌回收比值。
2.4.3 中和法 取“2.1”项下菌液0.1 ml,至9.9 ml“2.3.2”项下1:10供试液中,混匀,作为试验组;取“2.1”项下菌液0.1 ml,至9.9 ml“2.3.2”项1:20供试液中,混匀,作为试验组;取“2.1”项下菌液0.1 ml,至9.9 ml“2.3.2”项的1:50供试液中,混匀,作为试验组;取“2.1”项下菌液0.1 ml,至9.9 ml TSB中,作为菌液对照组,依法操作,计算各试验菌回收比值。
2.5 回收比值计算
采用平皿法或薄膜过滤法。
试验组回收比值=(试验组菌落数-供试品对照组菌落数)/菌液对照组菌落数,中和剂回收比值=(中和剂对照组菌落数-供试品对照组菌落数)/菌液对照组菌落数[15]。
2.6 结果判断
方法适用性试验中,若采用平皿法或薄膜过滤法时,方法适用性试验各试验菌回收比值应在0.5~2范围内,若在稀释液或培养基中加入中和剂或灭活剂,中和剂或灭活剂对照组回收比值应在0.5~2范围内[16]。
2.7 微生物计数方法适用性试验
2.7.1 常规法 按“2.3.1”项下,常规法测定3批样品5种菌株回收比值,平皿法计数结果见表1。
由表1可见,3批样品5种菌株回收比值均未完全在0.5~2范围内,其中,白色念珠菌在TSA和SDA中回收比值均为0,黑曲霉在TSA中回收比值为0。

表1 云南白药胶囊微生物限度检查方法适用性回收比值(常规法,n=3)
2.7.2 稀释法 选取“2.7.1”项下敏感菌株,按“2.4.2”项下,采用1:20,1:50供试液进行检查,平皿计数法结果见表2。

表2 云南白药胶囊微生物限度检查敏感菌回收比值(稀释法,n=3)
由表2可见,稀释倍数为1:50时,白色念珠菌在TSA和SDA中的回收比值均为0。稀释倍数为1:20时,黑曲霉在TSA中回收比值小于0.5;稀释倍数为1:50时,黑曲霉在TSA中回收比值在0.5~2范围内。
2.7.3 中和法初筛试验 选取“2.7.1”项下敏感菌株,“2.3.2”项下供试液,按“2.4.3”项下中和法进行试验,平皿计数法结果见表3。
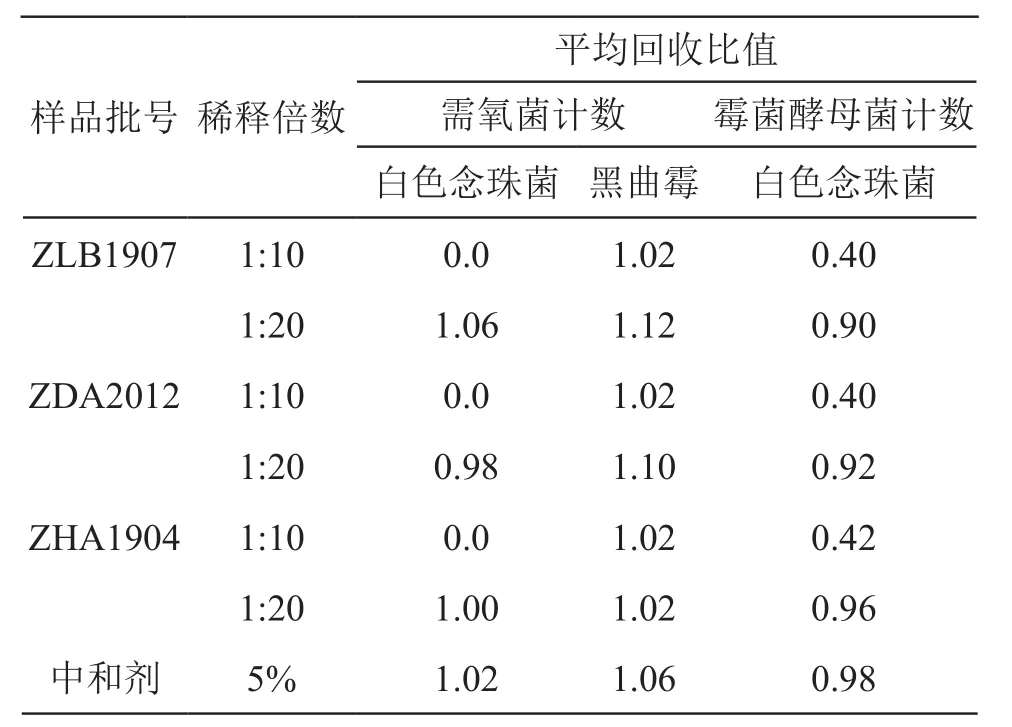
表3 云南白药胶囊微生物限度检查初筛试验回收比值(中和法,n=3)
由表3可见,稀释倍数为1:10,1:20时,黑曲霉在TSA中的回收比值均在0.5~2范围内;稀释倍数为1:20时,白色念珠菌在TSA和SDA中的回收比值均在0.5~2范围内。
2.7.4 中和法 取“2.3.2”项下1:20供试液,按“2.4.3”项下中和法进行试验,平皿计数法结果见表4。
由表4可见,稀释倍数为1:20时,各试验菌回收比值均在0.5 ~2范围内;中和剂各试验菌回收比值均在0.5 ~2范围内。
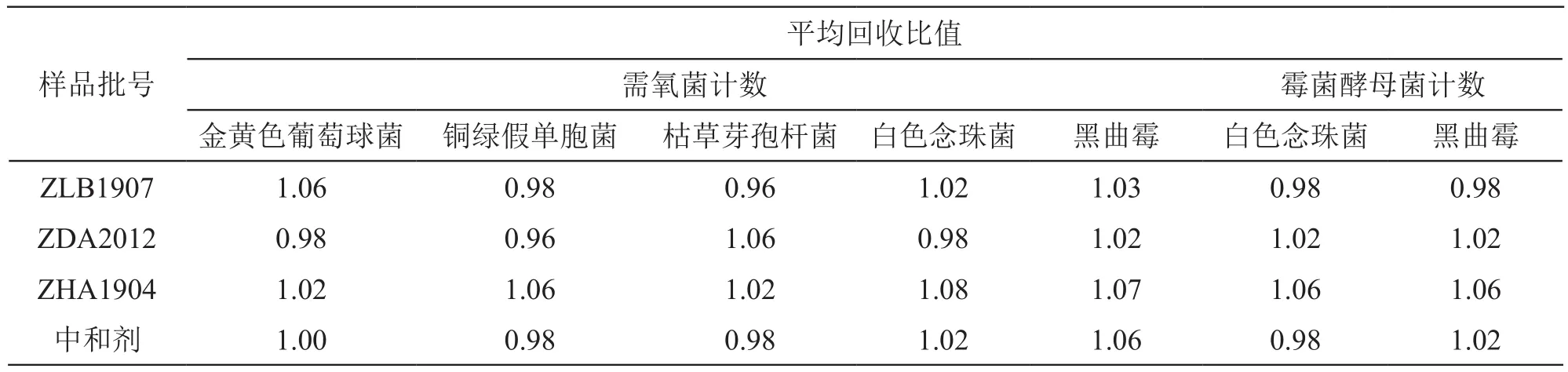
表4 云南白药胶囊微生物限度检查方法适用性回收比值(中和法,n=3)
2.8 控制菌检查
2.8.1 试验分组
2.8.1.1 耐胆盐革兰阴性菌 取“2.3.2”项下1:10供试液3组,每组1 ml,分别接种至10 ml EE中,一组作为供试品组,另两组分别加入不大于100 cfu的大肠埃希菌和铜绿假单胞菌,作为阳性对照组。
2.8.1.2 大肠埃希菌 取“2.3.2”项下1:10供试液两组,每组10 ml,一组接种至100 ml TSB中,作为供试品组,另一组加入不大于100 cfu的大肠埃希菌,作为阳性对照组。
2.8.1.3 沙门菌 取“2.2”项下供试品两组,每组10 g(至少含1粒保险子),分别接种至100 ml含5 %聚山梨酯80的TSB中,一组作为供试品组,另一组加入不大于100 cfu的乙型副伤寒沙门菌,作为阳性对照组。
2.8.1.4 金黄色葡萄球菌 取“2.3.2”项下1:10供试液两组,每组10 ml,分别接种至100 ml TSB中,一组作为供试品组,另一组加入不大于100 cfu的金黄色葡萄球菌,作为阳性对照组。
2.8.1.5 铜绿假单胞菌 取“2.3.2”项下1:10供试液两组,每组10 ml,分别接种至100 ml TSB中,一组作为供试品组,另一组加入不大于100 cfu的铜绿假单胞菌,作为阳性对照组。
2.8.2 控制菌检查结果 取“2.8.1”项下各组控制菌,照《中国药典》2020年版四部通则1106[17]依法检查,同时以稀释液代替供试液,其他同供试品组,作为阴性对照组,控制菌检查结果见表5。
由表5可见,中和法3批样品5种控制菌检查,阳性对照组均检出阳性菌,阴性对照组均无菌落生长,试验结果成立。结果表明,聚山梨酯80可消除药物抑菌性,中和法可用于云南白药胶囊控制菌检查。
3 讨论
微生物限度检查是药品安全性的重要指标,微生物实验的各项工作应在专属的区域进行,以降低交叉污染、假阳性结果和假阴性结果出现的风险。微生物限度检查应在不低于D级背景下的生物安全柜或B级洁净区域内进行[13]。本试验在D级背景下的生物安全柜内进行,避免了二次污染的风险,确保了检验结果的准确性、可靠性。
方法适用性试验的本质是建立对样品影响更小、操作率更高的方法[18],但其本身工作量大,且存在不同实验室方法欠佳、重复劳动过多的问题[19],去除药物抑菌性是其中的重点和难点[20],因此同一品种建立统一合理有效的方法适用性试验尤其重要。
由表1、表2可见,云南白药胶囊对白色念珠菌的抑菌性非常强,稀释倍数为1:50时回收比值仍为0,不适于继续采用稀释法。薄膜过滤法一般适用于抑菌性较强的样品,对于抑菌性非常强的样品,冲洗量太大,微生物易受损伤。为避免滤膜上微生物受损伤,降低微生物二次污染风险,建议采用中和法。
中和法由于稀释液和培养基中分别加入适宜浓度的中和剂[21-23],使样品在稀释或培养过程中抑菌性能得以彻底消除,适用于抑菌性强,稀释法、薄膜过滤法无法满足试验要求的样品的微生物限度检查。
中和法为《中国药典》2020年版四部通则收载的消除供试品抑菌活性的方法之一。聚山梨酯80为常用的中和剂,可用于钝化、中和供试品的抑菌活性,最好在稀释液或培养基灭菌前加入[24]。《中国药典》2020年版四部通则9202非无菌产品微生物限度检查指导原则中要求,如使用表面活性剂、灭活剂及中和剂,在确定其能否用于所检样品用量时,除应证明该试剂对所检样品的处理有效外,还需证明该试剂不影响样品中可能污染的微生物的检出(即无毒性)[25]。本试验在稀释液和培养基中分别加入了中和剂,中和剂各试验菌回收比值均在0.5 ~2范围内,确认中和剂有效和对微生物无毒性。该中和剂的加入,不影响微生物限度污染指示菌的检出。
云南白药胶囊为非无菌含药材原粉的中药制剂,按照《中国药典》四部通则1107的要求,耐胆盐革兰阴性菌应小于102cfu/g。本试验直接采用1:10供试液进行定性试验,当1:10供试液阳性对照组检出阳性菌时,表明该中和法可彻底消除该供试液中样品的抑菌性,随着稀释倍数的增加,样品的抑菌活性越来越弱,完全可以保障定量试验结果的准确性。该方法操作简单,工作效率高,适用于控制菌检查方法适用性试验初筛。
研究表明,云南白药胶囊为抑菌性较强的中药制剂,5 %聚山梨酯80可彻底消除药物的抑菌性,适用于云南白药胶囊微生物限度检查。
