蛋白精氨酸甲基转移酶5抑制剂研究进展
2021-07-08詹康宁黄张建赵立文
詹康宁,全 旭,黄张建,赵立文*
(1中国药科大学新药研究中心,南京210009;2南京圣和药业股份有限公司,南京210038)
蛋白质精氨酸甲基转移酶(protein arginine methyltransferase,PRMT)催化的精氨酸胍基甲基化,是一种常见的真核生物细胞翻译后修饰,影响细胞信号传导、基因转录、mRNA翻译、DNA重组和修复等多种生物学过程[1-4]。作为PRMT家族的成员,PRMT5介导的甲基化在维持正常细胞内环境平衡中发挥重要作用。然而,越来越多的研究表明,PRMT5的异常表达与多种肿瘤发生相关,目前已被认定为抗肿瘤药物研发的重要靶点[5]。本文介绍了PRMT5的结构、生化功能及其与肿瘤的相关性,对目前在研的PRMT5抑制剂进行综述,并讨论了PRMT5抑制剂的未来发展方向。
1 PRMT家族
PRMTs甲基化特定精氨酸胍基氮原子,由S-腺苷甲硫氨酸(S-adenosyl methionine,SAM)提供甲基,同时释放出一个分子的S-腺苷-L-同型半胱氨酸(S-adenosyl-L-homocysteine,SAH)。根据修饰形式,将PRMTs分成3类(图1):第1类(PRMT1、2、3、4、6、8)催化形成ω-NG-单甲基精氨酸(monomethyl⁃arginines,MMA)和ω-NG,NG-不对称二甲基精氨酸(asymmetric dimethylarginines,aDMA);第2类(PRMT5、9)催化形成ω-NG-MMA和ω-NG,N'G对称二甲基精氨酸(symmetric dimethylarginines,sD⁃MA);第3类(PRMT7)只催化ω-NG-MMA的形成[6]。PRMT1和PRMT5分别是主要的aDMA和sDMA形成酶,两者相较于其他PRMTs底物更多,目前研究表明在多种肿瘤中PRMT5表达水平升高[5]。

图1 3种PRMTs催化精氨酸甲基化反应
2 PRMT5的结构特征
PRMT5是细胞中主要的sDMA形成酶,N端为TIM桶状结构蛋白(TIMbarrel),C端是甲基转移结构域(图2)[7-8]。TIMbarrel与甲基转移酶复合体蛋白50(methylosome protein 50,MEP50)结合形成活性更高的复合体[9]。甲基转移结构域由底物结合位点和SAM结合位点组成,这两个结合位点通过一条狭窄的疏水通道(由Leu312、Phe327和Trp579形成)相连,使底物和SAM接近。活性部位有两个高度保守的谷氨酸残基(E435和E444)形成双E环。每个谷氨酸残基与底物精氨酸的胍基形成一对盐桥,从而允许甲基从SAM通过SN2过渡态转移到精氨酸残基上[10]。

图2 PRMT5:MEP50复合体的总体结构(PDB:4GQB)(A)和放大的H4R3底物肽结合槽(B)[11]
在一项对线虫PRMT5的突变研究中,将底物口袋中的保守残基之一Phe379变为蛋氨酸,可以实现H4R3同时进行sDMA和aDMA[7]。同样的,当人PRMT5中相应位置的Phe327突变也相应导致aDMA活性增强[7]。Phe327是PRMT5特有的残基,与sDMA功能密切相关,这可能是设计PRMT5特异性抑制剂时要考虑的一个重要因素。
3 PRMT5的生化功能
PRMT5广泛存在于细胞核和胞浆中,通过调控组蛋白及多种非组蛋白的甲基化修饰影响各种细胞过程。
PRMT5通过许多核质底物的甲基化起作用,包括组蛋白H2A、H3和H4[12],Sm蛋白[13],RAF蛋白[14]等等。组蛋白精氨酸sDMA被认为是一种阻遏标记,调节基因转录[12]。也有证据表明PRMT5通过Sm蛋白甲基化参与调节哺乳动物正确的mRNA结构性剪接[15]。PRMT5通过RAF蛋白甲基化调控RAS-RAF-MEK-ERK信号通路[14]。
部分非组蛋白也被鉴定为PRMT5的底物,在细胞功能调控中发挥重要作用[16-18]。例如,在DNA损伤时,PRMT5甲基化肿瘤抑制因子p53的3个精氨酸(Arg333,Arg335,Arg337),改变了p53与DNA的结合活性,进而触发p53控制的基因表达程序改变[17]。此外,PRMT5可甲基化转录因子E2F-1和NF-κB的P65,从而诱导目标基因的表达[19-20]。并且PRMT5也可与其他细胞蛋白结合,如高尔基体蛋白GM130、核糖体蛋白S10,分为维持高尔基体结构和核糖体正确组装[21-22]。
4 PRMT5与肿瘤的关系
最近几年,作为一个抗肿瘤药物靶点,PRMT5越来越引起科学界的关注。它在多种肿瘤中过表达,包括胶质母细胞瘤、肺癌、前列腺癌等,并在不同肿瘤中发生机制不同[5]。
例如,在临床上越来越多人胶质母细胞瘤病例的发生与PRMT5过表达相关[23]。研究发现,定位于肿瘤细胞胞浆的长非编码RNA,LINC00515与SNHG16,分别通过miR-16与miR-4518调节PRMT5高表达,招募并限制肿瘤抑制基因ST7与PTEN的表达,导致肿瘤发生[24-25]。相同的,PRMT5在人肺癌组织中显示高表达,在组织培养中,下调PRMT5的表达和使用PRMT5选择性抑制剂可明显抑制肺癌A549和H1299细胞的增殖。研究显示PRMT5是蛋白激酶B的上游调节因子,直接与蛋白激酶B共定位并相互作用,从而诱导肺癌细胞增殖[26]。在前列腺癌中,PRMT5通过表观基因激活前列腺癌细胞中雄激素受体(androgen receptor,AR)的转录来促进前列腺癌细胞的生长[27]。免疫共沉淀结果显示PRMT5与负责AR转录的主要转录因子Sp1、ATP依赖的染色质重塑剂BRG相互作用,诱导AR基因启动子区域的H4R3对称甲基化,激活并促进细胞生长。因此敲减PRMT5或用特定的PRMT5抑制剂会减少AR的表达,从而抑制AR阳性(而非阴性)的前列腺癌细胞生长[18]。
此外,由于代谢酶5-甲硫腺苷磷酸化酶(5'-methylthioadenonine phosphorylase,MTAP)基因与人类染色体9p21上的抑癌基因CDKN2A非常接近,因此在CDKN2A基因缺失的肿瘤中经常伴随MTAP缺失,是肿瘤中突变频率最高的基因之一。MTAP缺失使其底物甲硫腺苷(methylthio⁃adenosine,MTA)积累,MTA能选择性抑制PRMT5甲基转移酶的活性,并使其对进一步的PRMT5抑制更为敏感[28]。这表明PRMT5抑制剂可以成为MTAP缺失或低表达肿瘤患者的一种高度选择性的治疗方法。
5 新型PRMT5抑制剂
近年来,PRMT5作为肿瘤治疗的潜在靶点受到特别关注[11,16,29-34]。目前已有大量PRMT5小分子抑制剂报道,根据化合物是否占据SAM结合位点,可将其分为两类,SAM非竞争性抑制剂和SAM竞争性抑制剂,其中SAM非竞争抑制剂占据底物结合位点与底物竞争,而SAM竞争抑制剂则是占据SAM的结合位点。
5.1 SAM非竞争性抑制剂
5.1.1 EPZ015666 EPZ015666是第一个报道的具有细胞增殖抑制活性且可口服的PRMT5抑制剂,其IC50为22 nmol/L[35]。在一组5种髓细胞白血病(myeloid cell leukemia,MCL)细 胞 系(Z-138,Maver-1,Mino,Granta-519和Jeko-1)中,EPZ015666以浓度依赖的方式降低了SmD3甲基化水平和细胞增殖效应,在PRMT5基因敲除细胞中也观察到同样的SmD3降低。利用细胞热位移分析进一步确认EPZ015666与细胞内的PRMT5特异性结合。该化合物在小鼠体内表现出良好的药代动力学特性,静脉给药2 mg/kg后,AUC0-t为1 110 h·ng/mL,t1/2为1.38 h,分布体积为1.67 L/kg,血浆清除率为30 mL/(kg·min)。小鼠经口给予10 mg/kg,cmax为3 500 ng/mL,AUC0-t为3 847 h·ng/mL,t1/2为1.62 h,生物利用度为69%[36]。在一项Z-138皮下异种移植肿瘤小鼠模型的21 d体内药效学研究试验中,经口给予EPZ015666(200 mg/kg,po,bid),肿瘤生长抑制率接近95%[36]。

PRMT5:MEP50-SAM-EPZ015666共晶结构显示(图3),在SAM存在下,EPZ015666结合在肽结合部位,提示它是与肽底物(Ki=5±0.3 nmol/L)而非辅因子SAM发生竞争。为了找到EPZ015666对PRMT5高选择性的原因,进一步对晶体结构分析,发现EPZ015666与许多残基相互作用,在sDMA过程中起重要作用。特别是四氢异喹啉(THIQ)结构在这个过程中发挥重要作用:(1)THIQ的苯环与SAM之间存在阳离子-π相互作用;(2)THIQ环与PRMT5特有的残基Phe327形成潜在的π-π堆积作用,其他PRMTs在该位置的残基是Met[7];(3)THIQ位于由Leu319、Tyr324、Phe327和Trp579形成的疏水小口袋中,不利于极性官能团或体积大的基团;(4)THIQ的叔胺氮原子在水介导下与E435相互作用,影响整体催化过程。此外,EPZ015666的手性羟基和E444的侧链之间可能存在氢键相互作用,因此EPZ015666与之对应的立体异构体活性会有较大差异。EPZ015666的末端嘧啶基团也和Phe327、Phe580有π-π堆积作用。上述相互作用可能是EPZ015666对PRMT5的高亲和力和高选择性的原因[36]。

图3 EPZ015666与PRMT5:MEP50和SAM的共晶结构(PDB:4X61)[35]
5.1.2 GSK3326595 化合物GSK3326595保持了THIQ结构,PRMT5酶抑制活性IC50为6.2 nmol/L,该化合物分别通过调节人结肠癌细胞株SW480中p53与人套细胞淋巴瘤细胞株Z-138中p21的活性抑制SW480与Z-138增殖,诱导细胞周期阻滞。在Z-138皮下肿瘤异种移植小鼠模型中,GSK3326595有效抑制肿瘤体积(0.2~100 mg/kg,po,bid或50~200 mg/kg,po,qd)。同时该化合物呈现良好的药代动力学特性,静脉给药1 mg/kg后,AUC0-t为415 h·ng/mL,t1/2为1.42 h,分布体积为1.42 L/kg,血浆清除率为39 mL/(kg·min)。小鼠经口给予1 mg/kg,cmax为1 050 ng/mL,AUC0-t为3 450 h·ng/mL,t1/2为0.25 h,生物利用度为80.7%[37]。它具有良好的脑通透性和抗肿瘤效果,目前开展的两项Ⅰ期临床试验,用于评价其在治疗实体瘤、非霍奇金淋巴瘤(NCT02783300)和骨髓增生异常综合征、急性髓性白血病(NCT03614728)的安全性和临床疗效。

5.1.3 其他PRMT5非竞争性抑制剂 为了开发更有效的PRMT5抑制剂,研究人员将GSK3326595的2位侧链移至3位,得到了一系列活性化合物,如化合物1对PRMT5具有很高的抑制活性(IC50=26 nmol/L)[38]。在此基础上,默沙东公司利用环合策略获得了3,4-二氢-2,6-萘啶-1-酮类抑制剂,结果提示了手性羟基对PRMT5的氢键作用的重要性。经过分离得到了不同构型的小分子化合物2和化合物3,其中一个化合物的活性(IC50=10.84 nmol/L)明显强于另一个化合物(IC50=481.5 nmol/L)。有趣的是,在化合物2和化合物3的萘啶酮4位引入两个甲基得到了活性更高的化合物4(IC50=1.53 nmol/L)和化合物5(IC50=56.89 nmol/L),提示在这个位置可能还存在除了能够与Phe327和Phe580形成π-π堆积之外的其他的作用力,因此值得开展进一步的结构优化探索[39]。此外,阿古诺公司将THIQ替换成三环结构,得到一系列化合物,其中代表的化合物6与GSK3326595极为类似,PRMT5酶抑制活性IC50为10 nmol/L[40]。

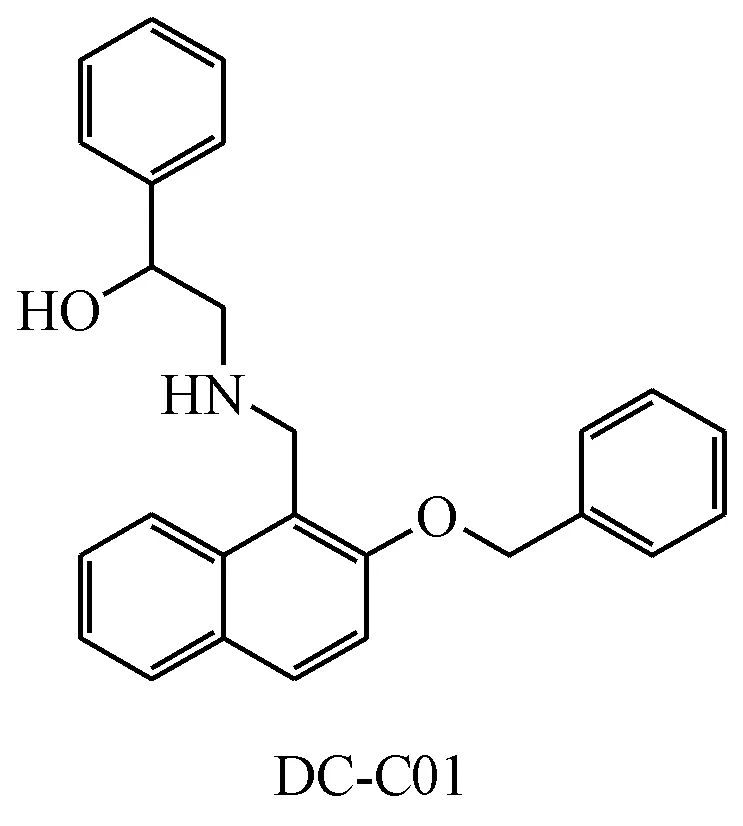
此外,研究人员在EPZ015666的基础上发现了一种新型萘环抑制剂,经过优化后得到了DCC01(IC50=2.8μmol/L),它能够呈剂量依赖地抑制一些肿瘤细胞(Z-138、Maver-1和Jeko-1)增殖[41]。分子对接结果显示,DC-C01的2位上苯环类似于EPZ015666的末端嘧啶,可能与Phe327和Phe580形成π-π堆积,而1位侧链上的苯环则可能与SAM形成阳离子-π相互作用,并且有可能与Phe327产生π-π堆积[41]。
5.2 SAM竞争性抑制剂
5.2.1 A9145C PRMTs含有高度保守的辅因子SAM甲基转移结合域,而SAH和天然产物西奈芬净则是SAM的结构类似物,两者均显示出良好的PRMT5抑制活性,但对其他PRMTs缺乏选择性。A9145C是早期基于西奈芬净结构研发出的SAM竞争抑制剂,在与PRMT5:MEP50和H4组蛋白肽段的晶体结构中显示,A9145C可以和SAM结合域中的几个残基(如,Asp419、Met420、Glu392和Try324)互相作用,但是它产生相互作用的结构与SAM对应的结构一致,包括腺苷部分的主体与两个氢键、尾部的羧基,仅有C5氨基部分可能与组蛋白H4产生额外的相互作用,因此它虽然能很好地结合在SAM结合域中,但仍然缺乏对PRMT5的选择性[9]。

5.2.2 LLY-283 在后续的研究中,MTA被证明是一种有效的、选择性的PRMT5抑制剂[28]。这一发现提示模拟SAM结构的核苷类似物有望对PRMT5产生较好的选择性抑制活性。为此,2018年,发现了一种有效的选择性PRMT5抑制剂LLY-283,具有较强的PRMT5酶抑制活性,IC50为20 nmol/L[42]。LLY-283是SAM的类似物,其中苯基取代了SAM中的蛋氨酸部分,且与核糖的构象十分类似。除了与相同残基的相互作用外,LLY-283的苯基与PRMT5特有的残基Phe327形成π-π堆积[42]。此外研究发现,将吡咯嘧啶换成其他芳杂环基团时,酶抑制活性下降[43]。在体外细胞增殖抑制实验中,LLY-283对在许多血液瘤和实体肿瘤细胞系(HCC1937、NUGC3、MV4-11、NCI-HI1930、A375等20余种)中都观察到了强大的抗增殖作用。在人黑色素瘤A375肿瘤异种移植小鼠模型中,给药28 d,LLY-283(20 mg/kg,qd)显著抑制肿瘤生长[42]。该化合物具有良好的ADME性能,小鼠经口给予10 mg/kg,cmax为3 646 ng/mL,AUC0-t为7 943 h·ng/mL,t1/2为3.41 h,生 物 利 用 度 为50%[42]。

5.2.3 JNJ6461978 JNJ64619178是第一个进入临床试验的基于SAM结构的模拟核苷类似物(NCT03573310),采用环戊烷替代了SAM中的四氢呋喃环。它对PRMT5酶的抑制活性IC50为6.3 nmol/L,并对 一 组 肺 癌 细 胞(A549、NCI-H441、H1048、HCC78)表现良好的增殖抑制活性[44-45]。分子对接显示,喹啉环与Phe327形成π-π堆积,并且可能占据了组蛋白底物精氨酸的位置,而喹啉上的氨基则与双E环中的E444相互作用。这种相互作用方式表明,JNJ64619178对SAM和底物都有潜在的竞争性作用[34]。

5.2.4 其他SAM竞争性抑制剂 LLY-283和JNJ64619178的发现激发了更多研究者对SAM竞争性抑制剂的兴趣。鉴于PRMT5的活性位点附近有一个特有的半胱氨酸C449,人们推测其可作为潜在的共价修饰位点[34]。最近,Prelude公司报道一个醛类化合物7,它和PRMT5的相互作用与LLY-283类似,所不同的是,该化合物的醛基能额外地与C449巯基产生亲核加成反应,在生理pH条件下,形成的中间体会自动消除一分子水,从而形成乙烯基硫醚(图4)。该化合物显示良好的PRMT5选择性,IC50为19.5 nmol/L[46]。研发共价抑制剂不仅有利于提高对化合物的选择性,或许还有助于将来解决PRMT5抑制剂潜在的耐药性。


图4 乙烯基硫醚的形成过程
6 总结与展望
近年来,PRMT5抑制剂已成为抗肿瘤药物研发的热点,然而目前已报道的大多数抑制剂仍局限于类似的骨架结构。在不同肿瘤发生过程中,PRMT5导致肿瘤发生的机制各有不同且部分尚未明确,为了更好地了解PRMT5的治疗潜力,还需要挖掘更多关于PRMT5功能的生物学信息。
对于SAM非竞争性抑制剂,诺华公司和Agios公司对该类抑制剂底物结合的模式进行了实验,发现EPZ015666能够有效地抑制许多细胞株的体外生长,但缺乏肿瘤细胞株选择性,并且它对肿瘤细胞的抑制能力与细胞中MTAP的状态无关,这就有可能在临床试验中转化为潜在的毒性[28,47]。在MTAP缺乏的细胞中,MTA累积并部分抑制细胞中的PRMT5,使肿瘤细胞对PRMT5抑制剂敏感。这可能提供一个新的结合思路,设计一种小分子协同MTA与PRMT5结合,并特异性结合MTA:PRMT5复合物,从而抑制肿瘤细胞生长。
总之,随着对PRMT5生化功能的进一步研究,各类化合物与PRMT5结合的共晶结构的不断被报道,安全的PRMT5抑制剂逐渐被人们所发现并最终应用于临床,使肿瘤患者获益。
