卒中患者肠道宏病毒组的组成和菌群特征
2021-07-08王展强徐开宇周宏伟
王展强,徐开宇,周宏伟
南方医科大学珠江医院检验医学部,广东 广州 510280
2016年全世界约有6.76千万例缺血性卒中病例,约2.7百万例死亡,给许多中低收入国家带来沉重的社会负担,影响卒中发病的危险因素主要有糖尿病、高血压、血脂紊乱等[1-2]。近年来越来越多的研究指出肠道菌群在卒中发病及预后中具有重要影响。与健康个体相比,卒中患者肠道菌群的多样性和活菌计数发生显著变化,菌群紊乱主要表现为机会病原体增加和有益菌群减少[3],并且独立于某些合并症(高血压、年龄和II型糖尿病)[3-4]。将卒中小鼠的肠道菌群移植给无菌小鼠[5]或将卒中患者的肠道菌群移植给抗生素治疗的小鼠中[6]均能加剧缺血引起的脑损伤面积和相关功能缺陷;缺血性损伤前服用广谱抗生素,小鼠卒中发生后预后明显恶化[7];此外,抗生素诱导的菌群失调还导致促炎性IL-17+γδT 细胞迁移和IL-17 相关趋化因子表达减少[8]。因此,肠道菌群可能通过调节肠道T细胞向脑部迁移来影响卒中后神经炎症的程度进而发挥作用。
人类肠道中定植有各种各样的病毒,这些病毒共同构成人类肠道宏病毒组,肠道宏病毒组与肠道菌群、真菌等共同构成肠道微生物群落。肠道宏病毒组中最主要的成员是能够侵袭细菌的病毒——噬菌体,其与细菌的比例约为1∶1[9]。在肠道微生物群落中,噬菌体丰度和菌群丰度的变化存在关联性,如在儿童发育过程中,肠道菌群的丰度与噬菌体的丰度呈负相关关系[10]。正常情况下肠道菌群和噬菌体处于平衡状态,当发生紊乱时,这种平衡状态被打破,这在疾病的发病过程中具有重要作用,如在炎症性肠病(IBD)患者中肠道宏病毒组的噬菌体丰度的改变与菌群丰度改变存在负相关关系[11]。
卒中患者的肠道菌群与健康志愿者存在显著差异,并且肠道菌群可能通过神经炎症反应发挥作用[7-8],目前尚无研究报道卒中患者肠道宏病毒组的特征,尚不清楚肠道宏病毒组中的噬菌体和肠道菌群之间捕食-被捕食关系在卒中发病过程中是如何影响彼此在肠道中的定植。本研究旨在探索卒中患者肠道宏病毒组的特征以及肠道宏病毒组与肠道菌群的相互影响。
1 资料和方法
1.1 纳入与排除标准
卒中组纳入标准:(1)年龄在18~80岁之间;(2)诊断为急性缺血性卒中;(3)大动脉粥样硬化性卒中亚型;(4)缺血性卒中发作后7 d内入院;(5)入院后48 h内收集粪便样本。
健康对照组纳入标准:(1)年龄在18~80岁之间无症状体检人群;(2)愿意提供新鲜的粪便样本。
排除标准:(1)入院前1个月内或入院后使用抗生素、益生元或益生菌;(2)卒中发作7 d后入院;(3)卒中发作后7 d内死亡;(4)与动脉粥样硬化无关的卒中(例如颈动脉夹层、心源性栓塞性卒中或其他TOAST 亚型);(5)全身疾病史,例如肝硬化、肾衰竭、肠道疾病(即IBD)和恶性肿瘤;(6)健康对照组志愿者无脑血管病史。
1.2 研究对象
选取2014年2月~2016年2月在南方医科大学南方医院神经内科就诊的急性缺血性卒中患者15名,同时招募15名年龄性别匹配的健康志愿者纳入研究。本研究经南方医科大学南方医院医学伦理委员会批准,所有志愿者均签署知情同意书。
1.3 方法
1.3.1 标本采集 对所有参与者,收集空腹EDTA抗凝血样本、新鲜粪便样本以及相关的临床数据。血液离心分离血浆后放在-80 ℃冰箱冻存;粪便样本收集后立即放在-80℃冰箱冻存。
1.3.2 病毒总DNA提取与检测 使用差速离心法富集粪便病毒颗粒(VLP)[12]:取粪便悬浮于无菌SM缓冲液[200 mmol/L NaCl,10 mmol/L MgSO4、50 mmol/L Tris-HCl(pH=7.5),0.01%gelatin]中并混匀,5000 g离心45 min;向样品中加入10%PEG 6000,4 ℃环境中放置12 h;4 ℃条件下15 000 g离心45 min,并将获得的沉淀重悬于SM溶液中,4 ℃环境中放置12 h;为了去除低分子量污染物,在室温下使用14 000 MWCO膜在SM缓冲液中透析样品(每2 h更换一次SM缓冲液);向同一离心管中依次加入2 mL的1.70 g/mL、1.50 g/mL和1.35 g/mL CsCl,加入4 mL含1.15 g/mLCsCl的样本,使总体积调整为10 mL,4 ℃条件下82 000 g离心2 h,收集密度介于1.36 g/mL~1.50 g/mL CsCl层的样本。采用病毒DNA(dsDNA和ssDNA)和RNA(ssRNA和dsRNA)共提取的方法提取核酸并使用Nano Drop检测核酸浓度和纯度。
1.3.3 肠道宏病毒组测序 使用超声破碎仪将核酸进行随机打断,将打断后得到的短片段DNA 用于构建测序文库,采用Illumina平台对质检合格的文库进行测序。
1.3.4 细菌总DNA提取与检测 使用MnikaGene Stool DNA Kit提取粪便总DNA,具体操作步骤参照说明书,使用Nano Drop检测总DNA浓度和纯度。
1.3.5 16S rRNAV4区基因片段的扩增及Illumina测序以粪便总DNA为模版,采用引物514F和805R[13](赛默飞世尔科技有限公司)。514F:GTGCCAGCMGCC GCGGTAA、805R:GGACTACHVGGGTWTCTAAT,用于扩增其中的16S rRNA V4区基因。PCR反应体系(20 μL):2×SYBR qPCR Master Mix(10 μL),上游引物(0.4 μL)、下游引物(0.4 μL)、RXO reference Dye2(0.4 μL)、模板(2 μL)、PCR水(6.8 μL)。PCR扩增条件:95 ℃预变性5 min,30个循环(95 ℃变性30 s,60 ℃退火30 s,72 ℃延伸30 s),72℃最终延伸5 min。采用Illumina Iseq100 平台对PCR 产物进行双末端100 bp测序。
1.3.6 病毒序列分析 使用Soapnuke[14]对原始测序数据进行质量评估并剔除低质量数据以得到高质量的clean data;使用BWA[15](v0.7.17,默认参数:mem-k 30)将clean data 与宿主数据库比对[宿主参考信息为人(Homo sapiens(human))],过滤比对长度低于reads总长80%的序列;使用Megahit[16](v1.1.2,默认参数:--presets meta-large-min-contig-len 300)对clean data 进行组装得到contigs,并使用blast(v2.9.0+)将contigs与宿主序列比对,去除宿主序列(判断标准:比对长度≥500 bp,比对相似性≥90%;比对长度占contigs 总长的80%以上,比对相似性≥90%);若样本数量大于1个,则使用CD-HIT[17](v4.7,默认参数:-c 0.95-aS 0.8)对所有样本的contigs进行聚类,获得unique contigs。同时使用基于参考序列的鉴定方法和Denovo病毒序列鉴定方法[18-21]对得到的unique contigs进行鉴定,将基于参考序列鉴定得到的确定病毒和Denovo病毒序列鉴定得到的新病毒序列作为最终纳入分析的病毒序列并注释物种信息。两种鉴定方法的鉴定标准如下:
(1)基于参考序列的鉴定方法:使用blast(v2.9.0+)将获得的unique contigs与病毒数据库(从NT数据库中分离)比对并进行筛选,筛选标准如下:A.比对相似性≥80%,比对长度≥500 bp,e≤1.0×10-5,可信度较高,定义为确定病毒(Confirmed virus);B.不满足A中的条件,但是比对长度≥100bp,e≤1.0×10-5,可信度低,定义为疑似病毒(Suspected virus),需要深入分析验证。
(2)Denovo病毒序列鉴定方法:①寻找候选病毒序列:将contigs与多个数据库比对,只需满足如下3个条件中一个即可:A.使用blastn(v2.9.0+)与分离自NT的病毒数据库(Virus-NT,包含噬菌体)比对,筛选e≤1.0×10-5的比对结果;B.使用blastx(v2.9.0+)与分离自NR的病毒数据库(Virus-NR,包含噬菌体)比对,筛选e≤1.0×10-3的比对结果;C.使用MetaGeneMark[22](v3.38)预测基因,然后使用hmmsearch[23](v3.2.1)将蛋白序列与HMM数据库比对(VPFs和vFam),筛选e≤1.0×10-5的比对结果;②排除假阳性:A.使用blast(v2.9.0+)将上述获得的候选病毒序列与NT数据库比对,筛选e≤1.0×10-10的比对结果;B.将前一步未比对上的序列与NR数据库diamond(v0.9.10)比对,筛选e≤1.0×10-3的比对结果;C.使用NCBI Taxonomy数据对前述比对结果进行注释,如果在前50个比对结果中,有20%以上的比对结果为非病毒序列(注释结果为Eukaryota、Bacteria、Archaea),则该序列被认为是非病毒序列,其余序列被认为是病毒序列。
1.3.7 病毒丰度统计 使用BWA[15](v0.7.17,默认参数:mem-k 30)将去除宿主序列后的clean reads与获得的病毒contigs比对,过滤比对长度低于reads总长80%的比对结果,统计病毒reads比例;计算每条病毒contig的RPKM值,计算公式如下:

Contig reads:比对上某contig 的reads 数,Total mapped reads:比对上的reads 总数,以百万为单位;Contig length:Contig长度,以KB为单位。
根据病毒contigs与virus-NT数据库比对结果,选择e≤1.0×10-5的best hit比对结果进行物种注释,统计病毒reads的分布。
1.3.8 噬菌体宿主预测 使用CRISPR Recognition Tool(CRT)从Refseq数据库的细菌基因组中构建CRISPRCas spacer数据库,然后使用blastn-short(v2.9.0+)与前述鉴定的病毒contigs比对,获取噬菌体可能的宿主信息[18,24]并用宿主信息对序列进行注释。筛选标准如下:e-value≤1.0×10-10,比对相似性≥95%。
1.4 统计学分析
使用SPSS、GraphPad Prism和R语言进行统计分析。参数资料采用均数加减标准差的方式进行描述并用配对设计t检验进行统计分析;非参数资料采用中位数与四分位数间距的方式进行描述并用配对资料的Wilcoxon检验进行统计分析;用Spearman检验特征菌的相对丰度和噬菌体相对丰度的相关性;根据单个样本病毒序列或细菌序列的相对丰度评估不同样本间的微生物群落结构差异(α多样性和β多样性);采用LEfSe分析(linear discriminant analysis coupled with effect size measurements)寻找两组间差异的特征病毒或细菌,用Mann-Whitney比较特征病毒或细菌在两组间的相对丰度。P<0.05(双侧)为差异有统计学意义。
2 结果
2.1 宏病毒组测序概况
本研究对每个样本进行10 G数据量的宏病毒组测序,下机数据过滤低质量数据和数据质控后剔除宿主序列,并使用Megahit组装成contigs,contigs长度主要集中在400~600 bp之间(图1A)。用基于参考序列的鉴定方法鉴定出1991种确定病毒(图1B),其中噬菌体和非噬菌体病毒各占83.12%和16.88%,鉴定出21 231种疑似病毒(图1B),其中噬菌体和非噬菌体病毒各占85.30%和14.70%;用Denovo病毒序列鉴定方法鉴定出5924种病毒(图1B),其中噬菌体和非噬菌体病毒各占69.38%和30.62%。因此纳入后续分析的病毒种类共6748 种病毒,其中噬菌体和非噬菌体各占73.49%和26.51%(图1C)。此外,对这些噬菌体序列进行物种注释时结果显示这些序列中的大部分属于微小噬菌体科(Microviridae)、短尾噬菌体科(Podoviridae)、肌尾噬菌体科(Myoviridae)和长尾噬菌体科(Siphoviridae)等病毒科(图2A)。

图1 宏病毒组测序概况Fig.1 Overview of gut virome sequencing.A:Contigs length distribution assembled by Megahit.B:Venn diagram of numbers of virus obtained from identification method based on sequence and Denovo identification method.C:Ratio of phages and non-phages in the sequence included in analysis.
2.2 卒中患者的肠道宏病毒组组成与健康志愿者存在差异
对上述纳入分析的序列进行注释和相对丰度统计,并分析卒中患者和健康志愿者之间肠道宏病毒组成和结构的差异,结果显示在科水平上,微小噬菌体科(Microviridae)、短尾噬菌体科(Podoviridae)、丝杆病毒科(Inoviridae)、肌尾噬菌体科(Myoviridae)和长尾噬菌体科(Siphoviridae)在卒中患者和健康志愿者肠道宏病毒组成中均占据优势(图2A)。两组间肠道宏病毒组的α多样性(P=0.320,图2B)和β多样性(P=0.169,R2=0.037,图2C)无显著差异,两组间肠道宏病毒组的结构相似;LEfSe 分析结果显示,在卒中患者肠道宏病毒组中,Cronobacter phage CS01和Bacteroides phage B40_8的相对丰度升高,而Human gut microviridae SH_CHD12和Streptococcus phage Javan249 的相对丰度则降低,两组间宏病毒组的组成存在显著差异(图2D)。
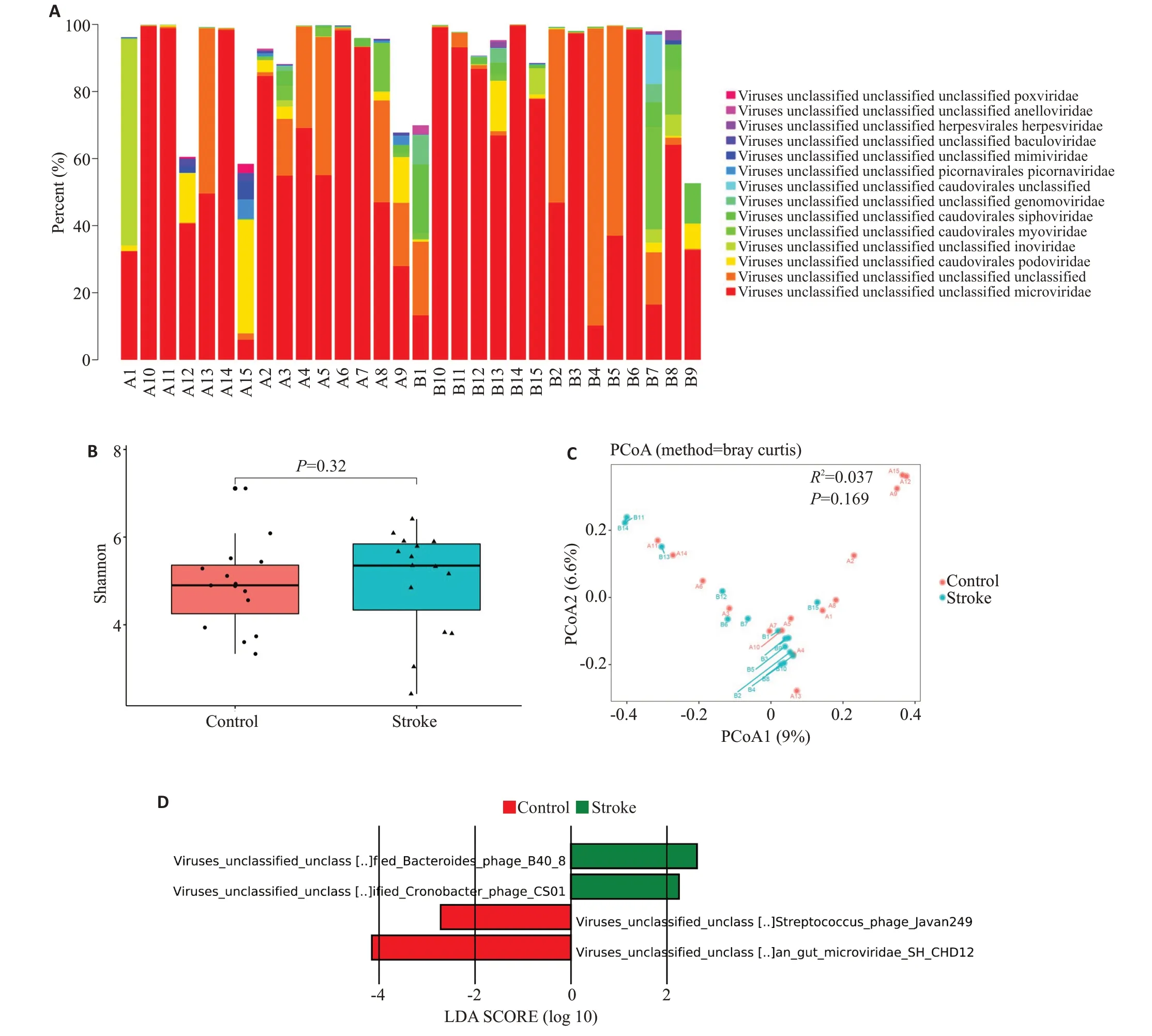
图2 卒中患者的肠道宏病毒组组成与健康志愿者存在差异Fig.2 Difference in virome composition between patients with stroke and healthy volunteers.A:Stacking diagram of virus at the family level of each sample.B:Difference in alpha diversity (Shannon index) of the virome between the two groups(P=0.320);C:Difference in beta diversity(Bray Curtis)of the virome between the two groups(P=0.169,R2=0.037).D:Results of LEfSe analysis.
2.3 卒中患者的肠道菌群与健康志愿者存在差异
16S rRNA测序结果显示两组间菌群的α多样性无显著差异(P=0.950,图3A),而β多样性则不同(P=0.005,R2=0.117,图3B),卒中患者和健康志愿者肠道菌群的结构存在差异。LEfSe分析结果显示在细菌属水平,卒中患者肠道菌群中Megasphaera等菌属的相对丰度增加,而Bifidobacterium等菌属的相对丰度则降低(图3C),与健康对照组相比,卒中组肠道菌群中益生菌的相对丰度下降,而机会致病菌的相对丰度增加。
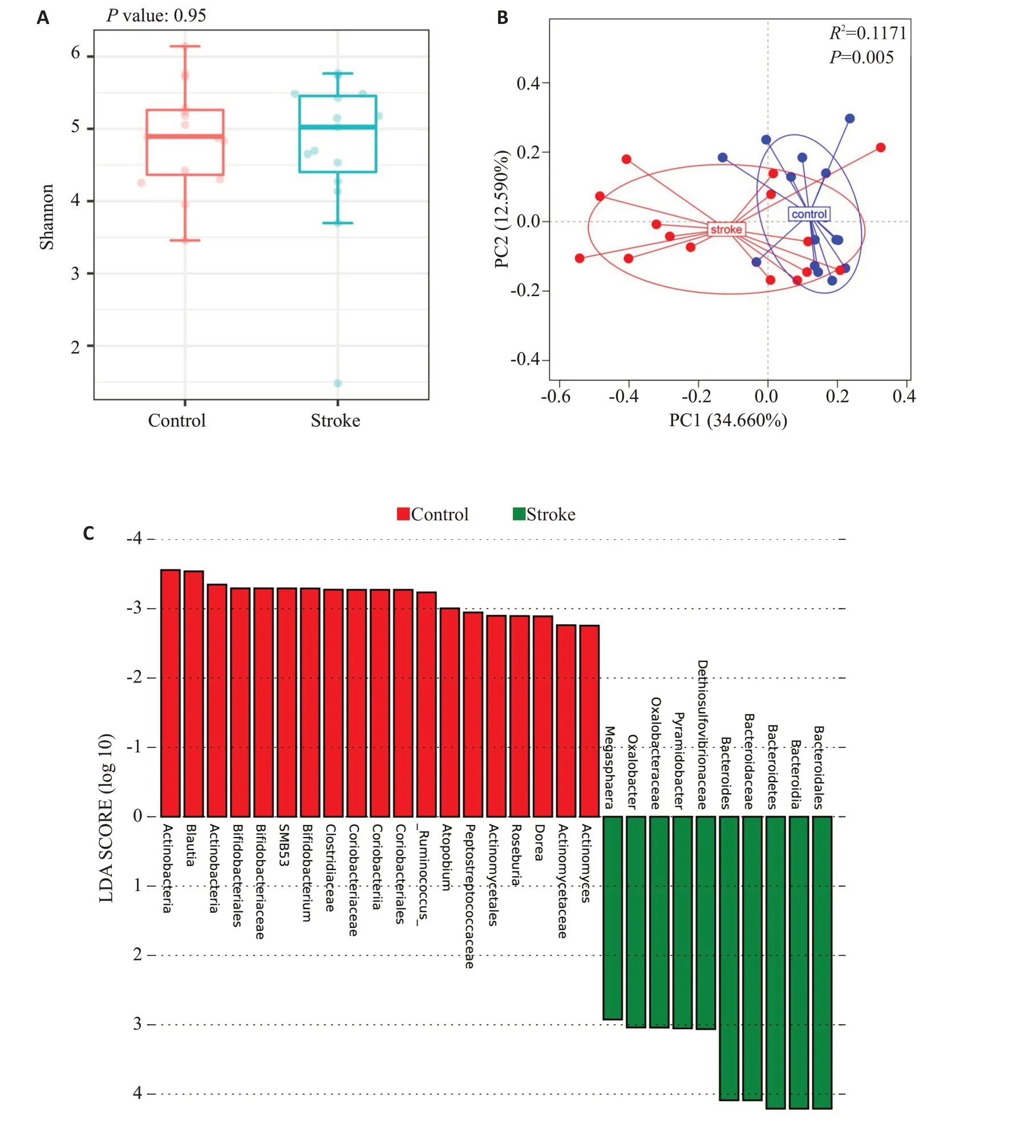
图3 卒中患者的肠道菌群与健康志愿者存在差异Fig.3 Difference in gut microbiome between stroke patients and healthy volunteers.A:Difference in alpha diversity (Shannon index) of microbiome between the two groups (P=0.950).B:Difference in beta diversity (weighted unifrac) of microbiome between the two groups(P=0.005,R2=0.117).C:Results of LEfSe analysis.
2.4 肠道菌群与宏病毒组存在相互影响
在鉴定为噬菌体的contigs中,共有576个contigs通过比对获得其宿主信息,获得宿主信息的contigs约占鉴定为噬菌体的contigs总数的11.62%。在肠道宏病毒组中只有Faecalibacterium和Bilophila两个菌属的噬菌体相对丰度在两组间具有差异(Faecalibacterium:P=0.001;Bilophila:P=0.048,图4A),并且这些噬菌体在卒中组的相对丰度均高于健康对照组。噬菌体与菌群相对丰度相关性分析显示,健康对照组中,Bilophila的噬菌体与其宿主、Faecalibacterium 的噬菌体与其宿主、Roseburia 的噬菌体与其宿主均呈正相关关系(Bilophila:r=0.541,P=0.040;Faecalibacterium:r=0.520,P=0.049;Roseburia:r=0.526,P=0.046,图4B);而卒中组中仅有Streptococcus的噬菌体与其宿主呈正相关关系(r=0.550,P=0.036,图4C)。

图4 肠道菌群与宏病毒组存在相互影响Fig.4 Interaction between microbiome and virome.A:Comparison of relative abundance of phages in the virome between the two groups(Bilophila:P=0.048;Faecalibacterium:P=0.001).B:Results of Spearman correlation analysis of relative abundance of phages in the virome and their hosts in the control group(Bilophila:r=0.541,P=0.040;Faecalibacterium:r=0.520,P=0.049;Roseburia:r=0.526,P=0.046).C:The results of Spearman correlation analysis of the relative abundance of the phages in the virome and their hosts in stroke group(Streptococcus:r=0.550,P=0.036).*P<0.05,**P<0.01.
3 讨论
人类肠道中定植有各种各样的病毒,这些病毒共同构成人类肠道宏病毒组。肠道宏病毒组中最主要的成员是具有DNA 性质的噬菌体,其与细菌的比例约为1∶1[9]。本研究采用高通量测序的方法对卒中患者和健康志愿者的粪便DNA病毒进行建库测序和宏病毒组分析。所获得的序列中有73.49%属于噬菌体序列,这与Virgin等[25]的研究结果相一致,提示噬菌体是肠道宏病毒组的重要组成部分,其中大部分噬菌体是有尾噬菌体目的成员。此外,目前已知的spacer序列有限,因此只有一部分病毒contigs能预测出可能的宿主,而在本研究中仅有11.62%contigs能够获得宿主信息。
病毒可能是肠道微生物组中多样性和丰度占优的物种[26]。多项研究指出病毒在多种疾病发病中具有潜在影响如IBD[11,27]、肥胖[28]等,在IBD 的患者中具有特异性改变[11]。本研究结果显示卒中患者和健康志愿者宏病毒组的整体多样性相似;但两者宏病毒组的组成不同,在卒中患者肠道宏病毒组中,Cronobacter phage CS01和Bacteroides phage B40_8的相对丰度升高,而Human gut microviridae SH_CHD12和Streptococcus phage Javan249的相对丰度则降低。
噬菌体是侵袭细菌的病毒,噬菌体和细菌间存在着“捕食与被捕食”的关系[29]。在肠道微生物群落中,噬菌体丰度和细菌丰度的变化存在关联性,如在儿童发育过程中,肠道菌群的丰度与肠道噬菌体的丰度呈负相关关系[10]。在正常情况下菌群和噬菌体处于平衡状态,当发生紊乱时,这种平衡状态被打破,这在疾病如IBD[11]的发病过程中具有重要作用。本研究发现健康志愿者和卒中患者的肠道宏病毒组与菌群相互作用的模式存在差异,在健康志愿者肠道宏病毒组中,除了共生菌的噬菌体与其宿主呈正相关外如Faecalibacterium的噬菌体与其宿主,机会致病菌的噬菌体的相对丰度与其相应宿主呈正相关,如Bilophila的噬菌体与其宿主、Roseburia的噬菌体与其宿主;而在卒中患者肠道宏病毒组中,仅是机会致病菌的噬菌体的相对丰度与其宿主呈正相关如Streptococcus的噬菌体与其宿主。
综上所述,本研究通过宏病毒组测序首次报告卒中患者肠道宏病毒组的特征,卒中患者肠道宏病毒组的整体多样性与健康志愿者相似,但是两者的病毒组成不同。此外,通过宏病毒组数据与菌群16S rRNA数据的相关性分析,本研究首次发现卒中患者和健康志愿者肠道宏病毒组与菌群相互作用的模式不同,在健康志愿者中共生菌和机会致病菌的噬菌体与其宿主呈正相关,而卒中患者中仅机会致病菌的噬菌体与其宿主呈正相关,提示卒中患者肠道内菌群与病毒群落的相互作用可能参与疾病进展。由于卒中患者肠道菌群处于长期紊乱状态,且与疾病进展存在关联[30],肠道宏病毒组是否可能成为卒中防治的新靶标,值得深入研究。
