孟德尔随机化模型及其规范化应用的统计学共识*
2021-07-07CSCO生物统计学专家委员会RWS方法学组
CSCO生物统计学专家委员会RWS方法学组
高 雪1 薛付忠2 黄丽红3 王 彤1△ 执笔 陈 峰4 夏结来5 主审
随机对照试验(randomized controlled trial,RCT)是评价因果效应的金标准,但由于受到伦理学、受试者依从性、研究期限等因素的制约,很多情况下难以实施。另外,RCT中纳入排除标准的限制可能导致研究样本与真实世界的人群出现异质性,因此研究结论的外推性也有待验证。相比之下,观察性研究和非随机对照研究数据更易获得,在样本的选择上也更接近真实世界的情况[1]。然而,观察性或非随机对照研究设计需借助恰当的因果模型来推断暴露因素(干预)与疾病结局之间的因果关联。
孟德尔随机化(Mendelian randomization,MR)是以遗传变异作为工具变量的统计模型。MR最早由Katan在探讨低血清胆固醇水平会直接增加癌症风险的假设中提出,近年来被广泛应用于因果关联研究中[2-4]。MR利用遗传变异在配子形成过程中随机分裂与组合的特性模拟对人群的随机分配过程:个体在出生时是否携带影响特定表型的遗传变异是随机的,而遗传变异在配子形成过程中既已确定,这一过程通常与后天的环境混杂因素是不相关的。因此,携带该变异与不携带该变异的人群在某结局上的差异则可以归因于暴露因素的变异,从而排除混杂因素的干扰[5](图1)。利用遗传变异作为工具变量的主要优势在于:遗传变异的形成独立于社会环境、生活习性和其他性状,这一特性保证了各个混杂因素在遗传变异的不同亚组间的均衡性,理论上避免了混杂因素的影响;另一方面,遗传变异的形成先于环境暴露、混杂及疾病结局的发生及水平变化,由遗传变异作为暴露的工具变量所解释的暴露变异也是先于结局的,由此排除了逆向因果问题[6]。
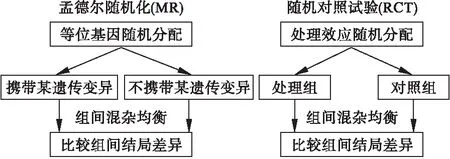
图1 孟德尔随机化和随机对照试验的比较
MR模型作为一种能够校正未测量混杂估计因果效应的统计学方法,在医学研究中得到广泛的应用,但针对模型的应用前提、核心假设、分析步骤以及结果解释等问题,还需进行一些必要的考虑与评价。为此中国临床肿瘤学会(CSCO)生物统计学专家委员会RWS方法学小组,经充分讨论,形成以下应用共识,以期促进MR模型的规范化应用和规范化报道。
MR模型的基本假设及常见的MR方法
1.MR模型的基本假设
遗传变异作为有效的工具变量需要满足以下核心假设(图2):
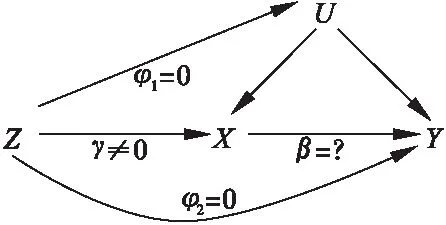
图2 工具变量核心假设Z:工具变量;X:暴露;Y:结局;U:混杂
(1)关联性:遗传变异Z与暴露X相关(γ≠0);
(2)独立性:遗传变异Z与影响“暴露—结局”关联关系的混杂因素U独立(φ1=0);
(3)排他性:遗传变异Z仅通过暴露X影响结局Y(φ2=0) 。
按照研究设计的不同,MR模型可以分为单样本MR模型与两样本MR模型。
2.单样本MR模型(one-sample MR)
单样本MR模型是指使用来自单个样本的数据构建MR模型,模型主要应用个体数据(individual data)作为研究样本,样本中同时包含每一个体的遗传变异、暴露以及结局的测量数据。
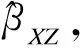
系数比估计量(比例估计值)的置信区间可通过正态近似法或Bootstrapping法得到。
此外,还可通过两阶段回归法得到因果效应的估计量。其中第一阶段由暴露X对工具变量Z进行回归,第二阶段由结局Y对第一阶段所得到的暴露估计值进行回归。当结局为连续变量且采用线性模型时,该两阶段回归法即为两阶段最小二乘法(two stage least square,2SLS),第二阶段回归时所得到的回归系数即为暴露对结局的因果效应估计值[7]。当使用多个遗传变异作为工具变量时,2SLS估计量可以看作各工具变量所对应的系数比估计量的加权平均,其中权重取各工具变量在第一阶段回归时与暴露的关联强度。选用系数比估计或2SLS估计要依据所拥有的样本数据情况来决定。当存在多个工具变量时,还可以将多个遗传变异整合为一个加权或非加权的等位基因得分(allele score),或称为基因风险得分(genetic risk score),再以得分作为工具变量构建MR模型[8-9]。
3.两样本MR模型(two-sample MR)
两样本MR模型中使用的遗传变异与暴露的关联关系统计量以及遗传变异与结局的关联关系统计量分别来源于两个独立的、不重叠的样本。模型主要应用汇总数据作为研究样本,样本数据中仅包含遗传变异与暴露及结局的关联关系汇总统计量(包括关联估计值、标准误、显著性P值等)。汇总数据一般来源于基于大样本的全基因组关联分析(genome-wide association study,GWAS),因此保证了两样本MR模型对因果效应的估计具有较高的效能,同时也提高了研究结果的可重复性。
MR模型的局限性及解决方法
应用满足核心假设的工具变量是MR模型得到有效估计的必要条件,然而,由GWAS中获取的绝大部分与暴露具有显著关联的遗传变异,并未完全掌握其关联的生物学机制,应用这些生物学机制尚不明确的遗传变异作为工具变量增加了其违背核心假设的可能。除工具变量核心假设外,不同类型的MR方法还有一些特定的前提假设,当假设违背时,将会得到有偏的、不一致的估计量。以下介绍MR模型在实际应用过程中面临的局限性问题,以及如何克服这些局限性,从而得到有效的估计量。
1.弱工具变量问题
应用强度高的工具变量可以有效提高MR模型估计因果效应的精度及效能,而当遗传变异与暴露关联性较弱时,会导致MR模型的估计量产生偏倚,称之为弱工具变量偏倚。当构建单样本MR模型时,估计量将会向受到混杂影响的观察性研究估计量的方向偏倚,且会造成Ⅰ型错误率(假阳性率)膨胀,当构建两样本MR模型且两个样本无明显重叠时,估计量将会向效应为零的方向偏倚[11]。
在MR模型中,常用Cragg-DonaldF统计量来评价工具变量的强度,弱工具变量所导致的偏倚大小和该统计量的期望值是相对应的。一个经验法则是工具变量强度F统计量至少为10,当F小于10时,因果效应的估计量会出现严重的偏倚[12]。另外,F统计量与“工具变量—暴露”的关联显著性P值是对应的,该P值通常作为工具变量的筛选阈值。为了保证模型中工具变量具有足够的强度,现有MR模型通常将全基因组显著性水平(P<5×10-8)作为阈值筛选工具变量,从而避免潜在的弱工具变量偏倚[12]。另外,当MR模型中工具变量的平均强度较弱时,还可以应用一些改进的MR模型,如基于修正权重的IVW,Egger-SIMEX等模型来校正潜在的弱工具变量偏倚[13-14]。
2.多效性问题
多效性是指遗传变异通过多种不同的路径对某一性状产生影响[15]。当利用多效性遗传变异作为工具变量时,相当于构造了“工具变量—暴露—结局”路径之外的其他通路,从而使得工具变量因违背核心假设而失效(图3)。应用存在多效性的工具变量会导致所研究的因果通路的效应估计产生偏倚,Ⅰ型错误率增加[16]。遗传变异的多效性效应是普遍存在的,而其对于MR模型的影响又是不可忽略的,因此如何排除多效性效应对于估计的影响是MR模型在实际应用中不可忽视的问题[17]。
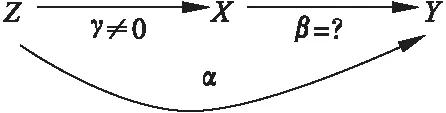
图3 多效性工具变量Z:工具变量;X:暴露;Y:结局
针对MR模型的多效性偏倚问题,目前的校正方法主要分为两类:第一类方法首先识别并排除存在多效性的工具变量,再利用剩余无多效性的工具变量构建MR模型进行因果效应估计。可借鉴识别离群点的思想来识别多效性工具变量,其基本思想是:基于有效工具变量得到的比例估计值应是同质的,比例估计值之间的差异应是随机的。此时多个比例估计值应基本分布在一条直线附近,而这条直线的斜率所代表的就是暴露与结局之间真实的因果效应值。反之,当某一工具变量对应的比例估计值与其他比例估计值之间存在显著的异质性时,则提示该工具变量违背核心假设。比例估计值之间的异质性主要来源于多效性工具变量,由于多效性效应被掺入因果效应通路中,导致对应的比例估计值产生偏倚,从而与其他工具变量所对应的比例估计值相比体现出显著的异质性[18]。
实际研究中,可以通过散点图、漏斗图等可视化方法结合统计检验方法识别多效性工具变量。常用的检验方法包含Q统计量检验[13,18]、MR-PRESSO异质性检验(mendelian randomization pleiotropy residual sum and outlier)[19]、HEIDI检验(heterogeneity in dependent instrument)[20]等。识别并剔除多效性工具变量后,可采用全局Q统计量检验、全局PRESSO检验、MR-Egger截距项检验等方法来评价剩余工具变量的多效性[21]。若剩余工具变量所对应的比例估计值已不存在异质性,则提示无多效性工具变量,接下来则可应用基于无多效性工具变量假设构造的MR模型进行效应估计[10,22]。
另一类方法为直接采用基于校正多效性偏倚的MR模型进行因果效应估计,这类模型允许工具变量存在多效性,在此情况下校正工具变量的多效性效应,同时估计暴露对结局的因果效应。基于校正多效性偏倚的MR模型中,基于个体数据的方法包含sisVIVE(some invalid some valid instrumental variable estimator)[23]、TSHT(two-stage hard thresholding)[24]、PRMR(pleiotropy-robust mendelian randomization)[25]等;基于汇总数据的方法包含MR-Egger[21]、基于中位数的估计(median-based estimate)[26]、基于众数的估计(mode-based estimate)[27]、CAUSE(causal analysis using summary effect estimates)[28]模型等。
具体研究中,常利用基于不同假设的MR模型进行敏感性分析,并比较各种方法所得结果的差异性,从而评估研究结果对于多效性假设的稳健程度[29]。不同方法所得到的估计量若是一致的,提示多效性工具变量所引入的潜在偏倚被有效的校正,而不同模型所得到的估计量若有显著的差异,则提示现有模型对于多效性工具变量较为敏感,有必要更进一步的进行分析与校正[30]。
3.连锁不平衡问题
连锁不平衡(linkage disequilibrium,LD)描述的是遗传变异之间的相关性,这种相关性通常是由遗传变异之间物理位置的临近所导致的。遗传变异之间存在LD时,每个遗传变异提供的信息不独立,当利用这些不相互独立的遗传变异作为工具变量时,则会导致效应估计产生偏倚。因此在构建MR模型时应尽量选择相互独立的遗传变异作为工具变量,而当工具变量间存在LD时,可应用纳入工具变量相关性信息的模型,如GSMR(generalized summary data-based MR)[22],从而避免连锁不平衡问题对于模型的影响。
4.人群分层问题
人群分层是指遗传变异与表型性状的关联性在不同种族或者国家的子群体中的异质性。这种异质性会导致遗传变异作为工具变量无法实现对于混杂因素的随机化过程,从而产生有偏的效应估计[31]。为了避免人群分层问题,在构建MR模型时通常选择同种族人群作为研究对象;在统计分析上,可以针对潜在的异质性因素进行分层分析,或利用主成分分析方法进行校正,从而排除由于人群分层问题导致的虚假关联出现[32]。
5.统计效能问题
MR模型估计因果效应的统计效能与纳入模型中的工具变量所解释的暴露变异比例相关,在样本含量一定时,工具变量对暴露变异的解释比例越高,模型的统计效能越高。因此,构建MR模型时要保证工具变量对于暴露变异的总体解释比例在一个较高的水平,从而保证模型具有充足的统计效能[33]。单个遗传变异对暴露变异的解释作用通常是很小的,因此现有MR模型通常选定多个遗传变异作为工具变量,从而增加工具变量对于暴露变异的解释比例,提高估计的统计效能。在模型构建上,Radial IVW[13]、Three-sample MR[34]、MRMix[35]等模型通过放宽工具变量与暴露关联的显著性阈值纳入更多的工具变量,同时校正由于弱工具变量的引入导致的潜在偏倚,从而提高模型的统计效能与估计精度。
样本含量是决定MR模型统计效能的另外一个主要因素。遗传变异的个体数据通常是难以获取的,并且由于研究经费限制,单个研究只包含较小的样本含量,因此基于个体数据的MR模型统计效能往往较低。相比之下,基于GWAS的汇总数据所构建的MR模型在数据获取,成本效率上具有更明显的优势,而数以十万甚至百万计的样本含量同时也保证了估计具有较高的统计效能。因此,目前研究大多利用汇总统计量构建MR模型,极大程度上推动了不同性状与疾病之间因果推断的研究。
另外,在选定工具变量构建MR模型进行因果效应估计之前,可以根据数据类型、样本含量、工具变量个数等指标选取适当的统计效能先验估计方法来预测模型的统计效能[36-38]。
6.结论外推问题
在对MR模型得到的因果效应估计量进行解释时,需要注意的一点是,遗传变异所解释的暴露变异只是暴露总变异中的一部分,因此利用遗传变异作为暴露的工具变量来估计其对结局的因果效应,所得到的效应值也只是由工具变量所决定的这一部分暴露变异对结局的效应,而由其他非遗传因素所决定的暴露变异对于结局的效应,是无法通过MR模型获得的。严格来说,由MR所得到的暴露对结局的效应估计量不能完全等同于真实的因果效应[39]。另外,在设计严谨,工具变量核心假设满足的情况下,MR模型可以为暴露与结局之间是否具有因果关联提供统计学上的线索,为后续更明确的试验研究及机制探索提供理论依据,但任何单一的研究方法都无法完全明确因果关系。真实的因果关联应结合疾病的生物学机制、完善的试验及临床研究结果等多方面证据综合进行探讨[40]。
MR模型的规范化应用报道
在实证研究中,构建MR模型进行因果效应估计主要包含以下步骤:根据研究设计选择合适的样本并收集数据;选择与暴露相关的遗传变异作为工具变量;根据样本数据的不同类型(个体数据、汇总数据)选择合适的MR模型进行估计与推断;根据统计分析结果做出客观的、合理的解释。上述步骤中需要注意的问题已在第二部分进行了详细的论述,总结来说,在构建MR模型进行实证研究时,需要对模型的假设及工具变量核心假设是否满足进行充分的评估,并对模型进行合理的选择与正确的应用,以确保统计结论的有效性。下面以结果检查报告表的形式给出MR模型的应用报道指南。

表1 MR模型结果报告检查表
