稻虾模式下克氏原螯虾组织及其养殖环境菌群多样性
2021-07-07王飞飞王夏雯金倩张智慧王信海
王飞飞,王夏雯,金倩,张智慧,王信海
(江苏省农业科学院宿迁农科所,江苏宿迁 223800)
克氏原螯虾 (Procambarusclarkii) ,俗称小龙虾,广泛分布于湖北、江苏、安徽、浙江、湖南、山东等省市。近年来,随着稻虾种养模式的推广,产业规模迅速增长,已成为中国重要出口创汇水产品之一[1],2017年小龙虾出口超过100万吨,销售额达到370亿美元,出口量占全球80%。随着养殖规模的扩大,病害问题也日益突出,如白斑综合征病毒病(White Spot Syndrome Virus,WSSV)、肠炎、甲壳溃烂病等[2-3],严重制约了克氏原螯虾养殖产业的健康发展。
微生物菌群在水产养殖中发挥重要作用,研究表明菌群与寄主健康有着密切的联系,如凡纳滨对虾感染WSSV后,肠道菌群组成和功能与健康虾相比差异明显[4],中华绒螯蟹感染肝胰腺病变综合征后,鳃、肝胰腺、肠道菌群变化幅度较大,且组织间菌群相关性明显[5]。另有研究表明水产品肠道等组织菌群结构与养殖环境密切相关[6-8]。稻虾共作模式是一种高效的种植与养殖相结合的生态系统,研究该模式下小龙虾肠道等组织及其养殖环境微生物多样性,有助于改善小龙虾养殖环境生态功能,提高养殖水平。目前相关研究未见报道。
高通量测序技术因其能完整反映测试样品的菌群结构特征,逐渐成为研究微生物菌群结构的重要工具[9],本试验基于Illumina Miseq测序平台对样品中的克氏原螯虾、肝胰腺、鳃、养殖水体以及池塘底泥等细菌微生物16S rRNA基因序列进行测定,分析稻虾共作模式下小龙虾肠道等组织及其养殖环境菌群结构,全面客观反应稻虾模式下小龙虾肠道、肝胰腺、鳃、水体、底泥微生物菌群多样性及其相互关系,为高效开展稻虾养殖及小龙虾疾病防控提供理论依据。
1 材料和方法
1.1 试验地点与样品采集
试验地点位于江苏省宿迁市宿豫区(33°97′32″N,118°32′30″E),属于亚热带气风气候,年均降雨量1 400 mm,采样地点稻虾共作已有5年,面积2.5 hm2,围沟宽5 m,深1.2 m,土壤为壤土。2019年7月2日放养平均质量为5 g的克氏原螯虾虾苗75 000尾/hm2,其间投喂专用配合饲料,每天上午、下午各1次。
采样时间为2019年8月20日上午 8:00—9:00,从池塘随机采集5尾大小相近的健康克氏原螯虾,平均体质量为(23.5±1.8)g,平均体长(9.5±0.2)cm。设置一个固定采样点,距离岸边2 m,分别用灭菌过的有机玻璃采水器和采泥器采集水样(水面下0.5 m)和底泥(水底表层5 cm),装入无菌采水瓶和采样袋。采集样品均置于冰盒中运至实验室。
稻虾共作水体 pH值的测量使用PHBJ-260便携式pH测定仪型(上海精密仪器厂生产);水温测定使用WNY-01直形棒式普通玻璃温度计,在水深0.5 m和1.5 m处分别测量取平均值;溶解氧采用YSI DO 200型便携式溶解氧测定仪(北京康高特科技有限公司生产);其余各项水化指标的测定均按照《水和废水监测分析方法》在实验室内完成[10]。水化分析采用《地表水环境质量标准》(GB3838—2002)和《渔业水质标准》(GB11607—89)的方法完成。
1.2 样品处理
样品运至实验室后,立即在超净工作台无菌环境下,对5尾小龙虾体表进行消毒、解剖,分别收集肠道内容物、肝胰腺、鳃,充分混合置于无菌离心管;取水样50 mL,用孔径为0.22 μm无菌纤维素滤膜过滤,过滤后,滤膜置于无菌离心管,底泥充分混合后置于无菌袋中。5份样品处理完成后立即放入干冰盒中,送至南京鑫普华生物科技有限公司进行样品处理及测序,具体测序方法、步骤按照文献[11-12]中的方法进行。
1.3 数据处理
测序后,根据序列首尾两端的Barcode和引物区分样品,并调整序列方向,Barcode允许的错配数为0,最大引物错配数为2;用Pandaseq软件,根据PE reads之间的Overlap关系,将成对Reads拼接(merge)成一条序列,最小Overlap长度为10 bp;拼接序列的Overlap区允许的最大错配比率为0.2,筛选不符合序列;用PRINSEQ软件,过滤reads平均质量值20以下的碱基,过滤掉N碱基数量大于5的序列;用Usearch软件,采用Denovo和Uchime结合的方式去除嵌合体,使用Gold数据库。
1.4 统计分析方法
1.4.1 OTU聚类与物种注释
将序列完全一样的Clean reads归为一种Tag,并统计每条Tag对应的丰度(即reads数目),将其中的Singletons(对应reads只有一条的序列)过滤掉,利用Usearch在0.97%相似度下进行聚类,对聚类后的序列进行嵌合体过滤后,得到用于物种分类的OTU(Operational Taxonomic Units)。从各个OTU中挑选出丰度最高的一条序列,作为该OTU的代表序列。使用Uclust方法,将该代表序列与已知物种的16S数据库(Silva数据库)进行比对,从而对每个OTU进行物种归类。
1.4.2 Alpha 指数分析
Alpha多样性(Alpha Diversity)是对某个样品中物种多样性的分析,包含样品中的物种组成的丰富度(Richness)和均匀度(Evenness)两个因素,通常用Observed species指数、Chao1指数、Shannon指数、Simpson指数以及PD_whole_tree指数等来评估某个样本的物种多样性。Observed species指数和Chao1指数反映样品中群落的丰富度(Species richness)。Shannon指数以及Simpson指数反映群落的多样性(Species diversity),PD_whole_tree指数反应了样品中物种对进化历史保存的差异,PD_whole_tree指数越大说明物种对进化历史保存的差异越大。Goods_coverage指数反应了测序的深度,指数越接近于1,说明测序深度已经基本覆盖到样品中所有的物种。
1.4.3 热图(Heatmap)分析
Heatmap是以颜色梯度来代表数据矩阵中数值的大小并能根据物种或样品丰度相似性进行聚类的一种图形展示方式。利用 R语言 Vegan包,Vegdist和Hclust进行距离计算和聚类分析;用chao算法计算距离,complete方法聚类,聚类结果加上样品的处理或取样环境分组信息,可以直观观察到相同处理或相似环境样品的聚类情况。
1.4.4 主坐标分析(Principal coordinates analysis,PCoA)
为了进一步展示样品间物种多样性差异,使用PCoA的方法展示各个样品间的差异大小。基于Bray curtis距离、Weighted Unifrac距离和Unweighted Unifrac距离来进行PCoA分析。
为了便于作图处理,稻虾共作模式下所取试验样品水体、底泥、肠道、鳃、肝胰腺分别用英文缩写CW、CS、CI、CG、CH代替。
2 结果与分析
2.1 水质主要理化指标
采集的稻虾共作水质指标如下:水温(27.6±0.1)℃,pH(8.11±0.05),化学需氧量(COD)为(23.31±1.8)mg/L,硝酸盐(NO3-N)(0.021±0.002)mg/L,亚硝酸盐(NO2-N )(0.005±0.001)mg/L,磷酸盐(PO4-P)(0.031±0.003)mg/L,TP(1.103±0.031)mg/L,TN(3.198±0.101)mg/L,各水质指标都在正常范围内。
2.2 测序结果
对稻虾共作各样品中高通量测序的结果进行统计(表1),在过滤掉低质量碱基序列和嵌合体后,5个样品共计获得有效序列393 519个,平均78 703.8,水体最多为94 687,肠道最少为48 659。为了保证后期分析结果合理,对每个样本的数据进行随机抽平处理,抽平数量按照最低样本数量进行抽平,抽平数量为48 659,将一致性在97%以上的序列聚类成一个OTU分类操作单位,5个样品抽平后共计聚类 7 223个OTU,平均1 444.3,底泥样本最多为2 064,鳃最少为831,测序深度99.25%~99.78%,说明试验样本中序列没有被检测出的概率较低。Alpha多样性发现Chao1指数、Observed-sopecies指数由高到低顺序依次为底泥>肝胰腺>肠道>鳃>水体,Simpson、Shannon指数由高到低顺序依次为底泥>肝胰腺>肠道>水体>鳃。说明底泥细菌丰度和多样性最高,水体细菌丰富度最低,鳃菌群多样性最低。
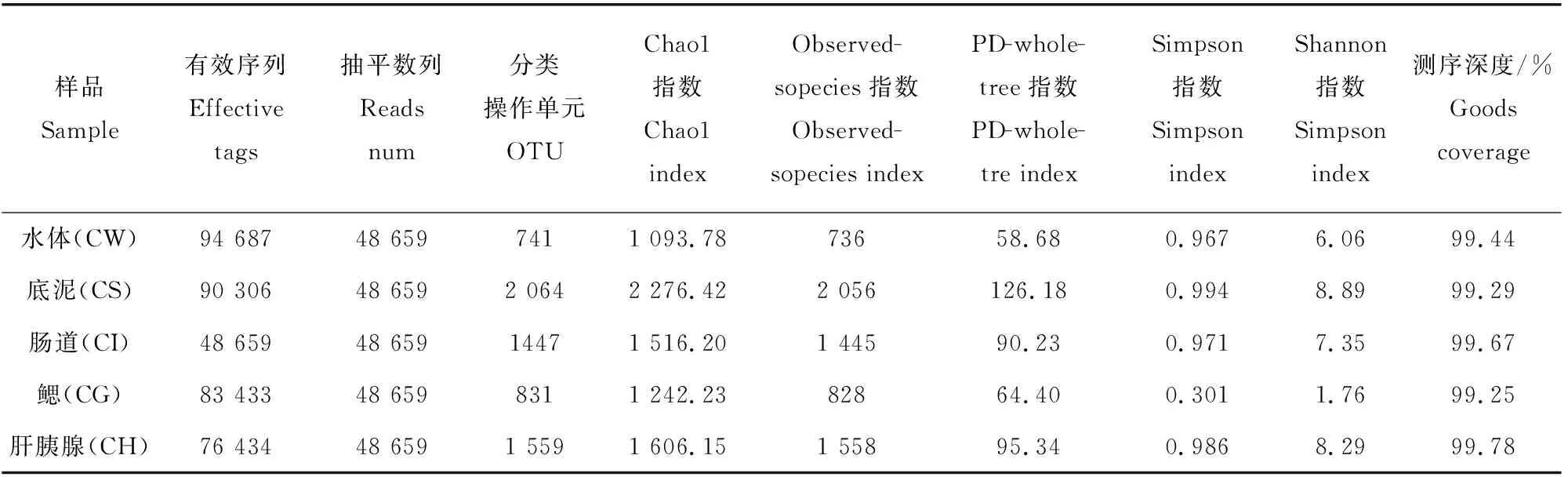
表1 样品测序数列及Alpha多样性分析
2.3 稻虾共作模式下各样品菌群组成分析
在门分类水平上,选取最大丰度前20个物种进行分析(图2),发现各样品菌群主要为变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和放线菌门(Actinobacteria)。变形菌门是底泥和鳃的优势菌门,是水体次优势菌门,占比均较大,其中鳃占比超过90%;厚壁菌门是肠道和肝胰腺优势菌门,占比均超过40%;放线菌门是水体优势种群,但在其他样品中占比较少。
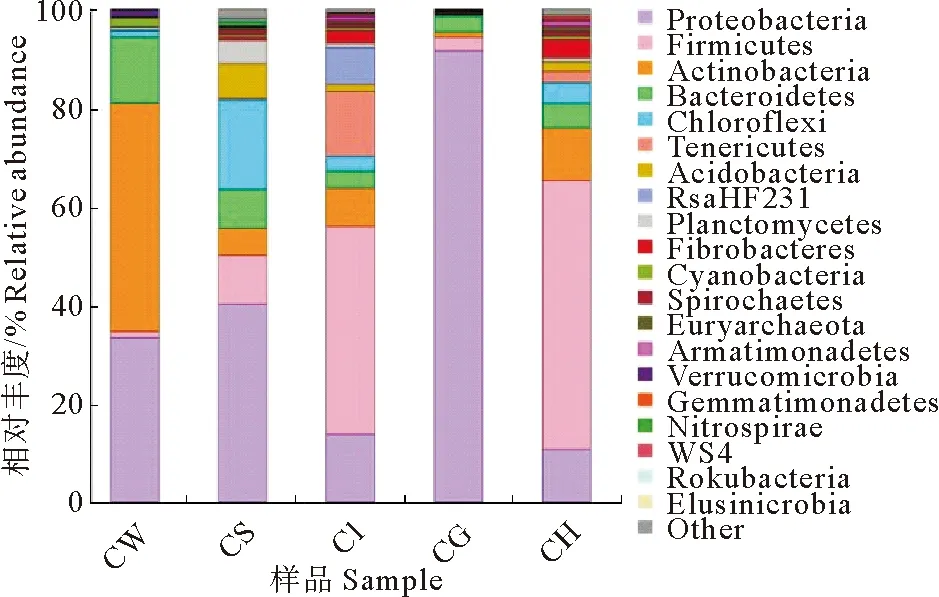
图2 基于门分类水平的菌群相对丰度分析
在属分类水平上(表2),水体、底泥、肠道、鳃、肝胰腺5个样品优势种群分别为放线菌门的hgcIclade(32.1%)、厚壁菌门的芽孢杆菌属(Bacillus2.1%)、柔膜菌门的CandidatusBacilloplasma(11.8%)、变形菌门的红育菌属(Rhodoferax87.2%)和厚壁菌门的芽孢杆菌属(Bacillus12.9%),各优势种群在样品中占比范围2.1%~87.2%,差异较大。底泥和肝胰腺含有共同的优势菌群芽孢杆菌属,芽孢杆菌属也是肠道、鳃的次优势菌群。厚壁菌门的Ruminiclostridium1在肝胰腺(6.5%)、肠道(3.7%)、底泥(1.0%)、鳃(0.3%)中作为主要菌群大量存在。

表2 测试样品基于属水平上的优势种群
根据 5个样品的微生物组成和相对丰度进行物种热图分析(图3),更加直观显示不同样品优势菌群之间差异,鳃和水体优势菌群优势明显。水体中的主要菌群变形菌门的Polynucleobacter、Limnohabitans(6.77%、6.51%),放线菌门的CandidatusLimnoluna(5.59%),变形菌门的OM43clade(3.41%)在其他样品中含量较少。
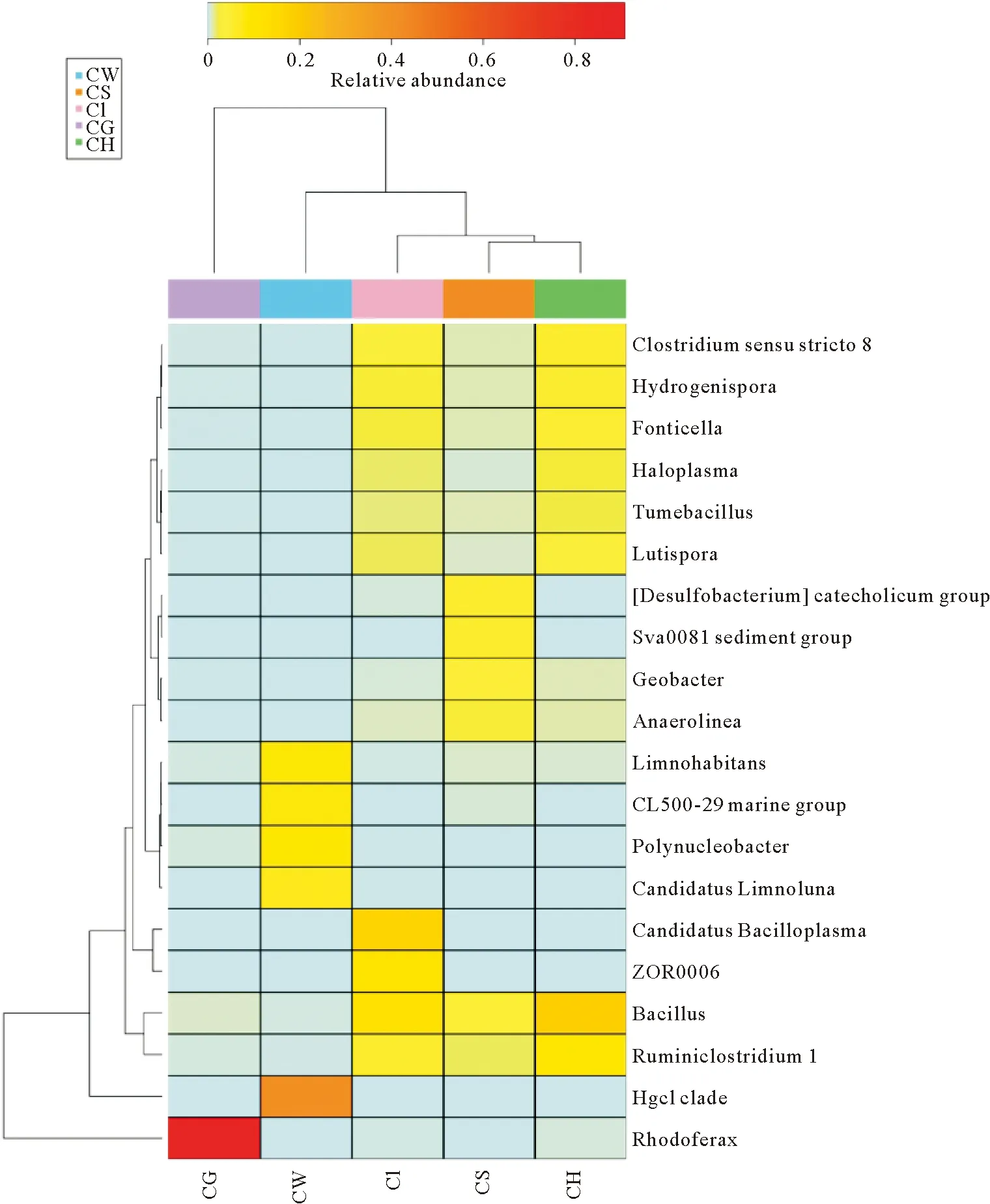
图3 属水平不同样品微生物丰度聚类热度
2.4 稻虾共作模式下不同样品间菌群相似性分析
根据OTU聚类分析结果,为了直观表现不同样品之间OTU数目组成的相似性及重叠情况,构建韦恩图(图4),5个样品共有248个OTU,占总数8.5%。肠道与肝胰腺、底泥、鳃、水体共有OTU个数分别为1 085、1 065、607、447,说明肠道与肝胰腺菌群关系最大,肠道菌群受到外界环境中底泥影响最大,底泥、肝胰腺、水体、肠道、鳃独有的OTU分别为637、247、116、108、27。说明底泥菌群更复杂,肠道次之。
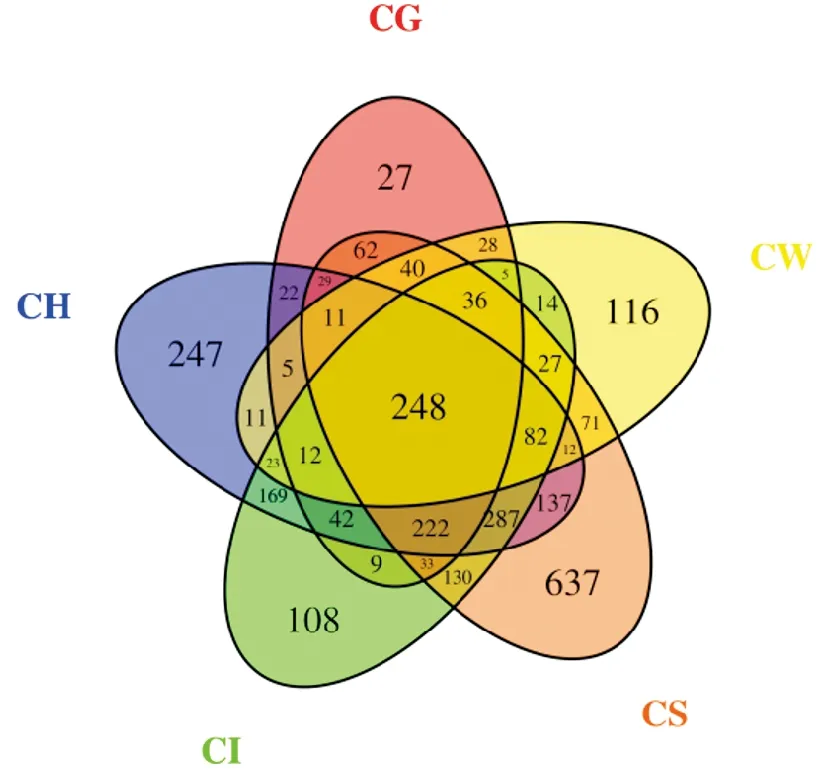
图4 各样品OTU 数量Venn分析
为了进一步展示样品间物种多样性差异,使用主坐标分析的方法展示各个样品间的差异大小(图5)。在稻虾共作模式下,底泥、肝胰腺、肠道相似度较高,其中肝胰腺和肠道菌群结构相似度最高,水体、鳃与其他样品微生物群落结构相似度较低。

图5 PCoA分析图
3 讨论
在水产养殖中,水产品体内定植菌群多样性与其功能息息相关,其中的优势菌群更是发挥着决定性的作用,是维护菌群平衡、保持宿主健康的重要因素[12],菌群结构及其多样性是养殖生态系统平衡与否的重要指标。稻虾共作模式因其具有良好的生态和经济效益,近年来得到广泛的推广与应用,本研究通过16S rRNA基因进行高通量测序,研究稻虾共作模式下克氏原螯虾肠道等组织及其养殖环境菌群多样性。结果显示:稻虾共作模式下稻虾田水体、底泥和克氏原螯虾肠道、鳃、肝胰腺5个样品共检测到有效序列393 519,抽平后共计聚类于 7 223分类操作单元OTU,测序深度均达到99%以上,水质指标检测结果都在正常范围内,说明本研究结果在一定程度上反映稻虾共作正常养殖情况下克氏原螯虾肠道、鳃、肝胰腺及养殖环境的菌群结构特征。
3.1 稻虾共作模式下菌群多样性分析
通过分析样品Alpha多样性指数可知,稻虾共作模式下,底泥的丰富度和多样性最高,这与拟穴青蟹[13]、凡纳滨对虾[4]、刺参[8]等水生动物的相关研究结果一致。叶建勇等[14]对精养塘克氏原螯虾肠道及其养殖环境研究显示底泥、水体菌群多样性高于肠道菌群,本研究中水体菌群多样性却远低于肠道等组织,这可能是因为稻虾共作模式下水稻大量吸收水体里的营养元素,水体清瘦,导致微生物数量较少。
3.2 稻虾共作模式下菌群组成分析
稻虾模式下5个样品菌群主要门类为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、柔膜菌门(Tenericutes)、厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)等,这与甲壳类水产品研究结果一致[4-5]。其中厚壁菌门是肠道、肝胰腺优势种群,是底泥、鳃第三优势种群。变形菌门是底泥和鳃优势菌群,是肠道、肝胰腺、水体次优势菌群。研究表明:厚壁菌门和变形菌门深度参与甲壳类肠道的生理和生化功能[15],厚壁菌门参与动物肠内多糖代谢,因此大多数食肉动物的粪便中厚壁菌门最为丰富[16]。与精养池单一饲料投喂相比,稻虾模式可为小龙虾提供丰富的昆虫和种子,因此本研究克氏原螯虾肠道厚壁菌门和变形菌门最丰富,与食肉动物肠道菌群成分类似。在属水平上进一步分析,CandidatusBacilloplasma是肠道优势菌属,研究表明:该菌属属于柔膜菌纲一个新家系[17-18],在甲壳类肠道菌群中大量存在并具有重要作用,目前对该菌属功能研究较少。在对中华绒螯蟹肝胰腺“白化”症体组织微生物多样性分析发现[5],患病的肝胰腺和肠道柔膜菌纲菌群大量繁殖且优势明显,但是水体底泥、鳃变化不明显,属于内发增值,柔膜菌纲的CandidatusBacilloplasm菌群如何变化尚不明确。陈一铭等[19]对感染白斑综合征(WSSV)克氏原螯虾肠道菌群分析发现,CandidatusBacilloplasm显著降低,可见CandidatusBacilloplasm菌群是克氏原螯虾体健康与否重要指标。底泥、肠道、肝胰腺含有共同优势种属Bacillus、Ruminiclostridium1,进一步说明底泥菌群对克氏原螯虾肠道、肝胰腺菌群影响较大。
3.3 稻虾共作模式下菌群相关性分析
韦恩图和PCoA结果显示底泥、肠道和肝胰腺菌群相关性较强,说明养殖环境中的底泥对克氏原螯虾肠道等组织菌群多样性影响较大。相关研究结果也显示养殖环境对水生动物肠道等菌群结构关系较密切,如王贤丰等[20]研究发现拟穴青蟹肠道、底泥和水体含有共同优势菌群,李存玉等[21]研究发现池塘和工厂化两种模式养殖的牙鲆肠道菌群均受水环境菌群影响。本研究结果也说明,养殖环境尤其是底泥对稻虾模式下克氏原螯虾肠道等菌群结构影响较大。本研究发现肠道和肝胰腺菌群结构相似性最大,说明二者菌群关系密切。有关研究发现[5],蟹肠道菌群的改变显著影响肝胰腺和鳃的菌群结构;感染WSSV的克氏原螯虾肠道菌群结构发生变化[2],进而影响肠道和肝胰腺功能发生变化;患有细菌性疾病克氏原螯虾肠道、肝胰腺均含有大量致病细菌[22]。这些研究结果也印证了肠道与肝胰腺菌群关联较强,但是二者相互影响的机理尚不清楚。
