反相高效液相色谱法测定阿立哌唑中的有关物质
2021-07-03尹霞陈斌王燕清徐朋陈渺丽徐慧娟吕林艳顾玲玲丽珠医药集团股份有限公司广东珠海519000
尹霞,陈斌,王燕清,徐朋,陈渺丽,徐慧娟,吕林艳,顾玲玲(丽珠医药集团股份有限公司,广东珠海 519000)
阿立哌唑(aripiprazole)是由日本大冢(Otsuka)公司与美国百时美施贵宝(Bristo-Myeres Squibb)公司共同研发的首个第三代非典型抗精神病药物,并于2002年11月15日经美国FDA批准上市,其化学名为7-{4-[4-(2,3-二氯苯基)-1-哌嗪基]丁氧基}-3,4-二氢氯喹酮,结构式见图1。阿立哌唑主要通过对多巴胺D2受体和血清素5-HT1A受体的部分激活,对5-HT2A受体的全部拮抗活性起效,临床主要用于治疗各种精神分裂症和情感障碍[1-4]。目前已经有关于阿立哌唑的有关物质测定的文献报道[5-8],且阿立哌唑的质量标准《中国药典》、美国药典和英国药典均有收载,但存在一些不足,比如未考虑到各杂质与主成分的响应是否有差异而笼统地采用主成分自身对照法控制各杂质的含量、流动相中采用高达25%的四氢呋喃(长期使用会腐蚀液相系统,降低色谱柱寿命)、流动相采用离子对试剂(延长平衡时间且降低柱寿命)、供试品溶液浓度较低灵敏度不够而导致杂质漏检、被检测杂质种类较少而影响产品质量的可控性和安全性、个别杂质在色谱柱上保留过强不出峰而漏检等。根据阿立哌唑的化学性质、合成路线及可能的降解途径,并通过强制降解试验考察,本研究以乙腈-0.05%三氟乙酸作为流动相,建立了同时测定阿立哌唑中7个杂质(A~G)的反相高效液相色谱(RP-HPLC)方法,并对特定杂质(E、F、G)进行校正因子研究。各已知杂质以及各破坏条件下产生的未知杂质在此条件下均能达到有效分离,可用于阿立哌唑的质量控制。
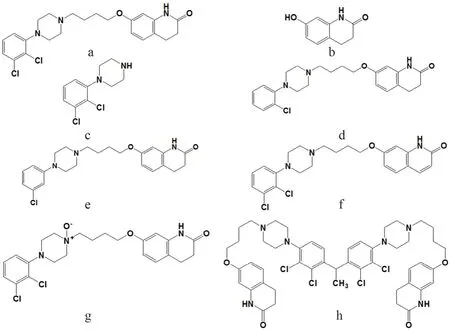
图1 阿立哌唑(a)及杂质A(b)、B(c)、C(d)、D(e)、E(f)、F(g)和G(h)的化学结构式Fig 1 Structure of aripiprazole(a)and impurity A(b),B(c),C(d),D(e),E(f),F(g),and G(h)
1 材料
1.1 仪器
Agilent 1260高效液相色谱仪,配备DAD检测器(安捷伦科技有限公司);Water 2697高效液相色谱仪,配备UV检测器(沃特世科技有限公司);梅特勒-托利多XS105微量天平(梅特勒-托利多国际股份有限公司);KQ-400DB数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药
阿立哌唑(批号:AP20716001、AP20716002、AP20818001,Neuland Laboratories Limited);阿立哌唑对照品(批号:100776-201302,含量:99.9%,中国食品药品检定研究院);杂质F(批号:ID 0042M2、ID 003LM7,含量:100%,EP);杂质G(批号:1805-073A6,含量:99.2%,TLC);杂质A、B、C、D、E(批号分别为160504、160326、140527、140526、160415,含量分别为99.1%、98.3%、99.0%、98.6%、99.0%,Molcan Corporation);乙腈、三氟乙酸为色谱纯;过氧化氢、盐酸、氢氧化钠为市售分析纯。
2 方法
2.1 色谱条件
采用Agilent ZORBAX SB-C18(150 mm×4.6 mm,5 µm)色谱柱;以乙腈-0.05%三氟乙酸(10∶90,V/V)为流动相A,以乙腈-0.05%三氟乙酸(90∶10,V/V)为流动相B;流速为1.2 mL·min-1;检测波长为254 nm;柱温30℃;进样量20 µL;按表1的程序进行梯度洗脱。

表1 梯度洗脱程序Tab 1 Gradient elution procedure
2.2 溶液配制
2.2.1 供试品溶液 精密称取本品约15 mg,置于50 mL量瓶中,加15 mL乙腈,超声使溶解,再加稀释剂(0.05%三氟乙酸)定容至刻度,作为供试品溶液。
2.2.2 自身对照溶液 精密量取供试品溶液0.5 mL,置已加入15 mL乙腈的50 mL量瓶中,用稀释剂定容至刻度,作为对照溶液。
2.2.3 空白溶液 以乙腈-稀释剂=3∶7作为空白溶液。
2.2.4 混合杂质对照品储备液 取7个已知杂质对照品各约3 mg,精密称定,置50 mL量瓶中,加乙腈适量超声使溶解,再加稀释剂定容至刻度,得到质量浓度约为60 µg·mL-1的单个杂质对照品溶液。精密吸取上述溶液各2.5 mL,置同一50 mL量瓶,加稀释剂定容至刻度,即得混合杂质对照品储备液。
2.2.5 阿立哌唑对照品储备液 取阿立哌唑对照品约30 mg,精密称定,置10 mL量瓶中,加入乙腈适量,超声使溶解,再加稀释剂定容至刻度,得到质量浓度约为3 mg·mL-1的阿立哌唑对照品储备液。
2.2.6 混合对照品溶液 精密量取上述混合杂质对照品储备液和阿立哌唑对照品储备液各1 mL,置10 mL量瓶,加稀释剂定容至刻度,即得。
2.2.7 系统适用性溶液 分别取阿立哌唑原料、杂质E和杂质F对照品适量,加乙腈适量超声使溶解,再加稀释剂配制成含阿立哌唑0.3 mg·mL-1、杂质E和杂质F各约1 µg·mL-1的混合液,摇匀,作为系统适用性溶液。
2.3 专属性试验
2.3.1 系统适用性试验 精密量取“2.2”项下空白溶液和混合对照品溶液各20 μL,按“2.1”项下色谱条件测定,记录色谱图,见图2,可知各杂质分离良好且阿立哌唑与杂质E、杂质F的分离度均大于2.0。
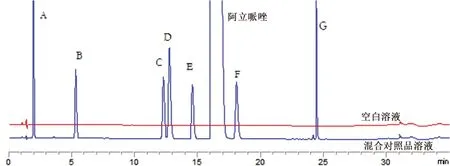
图2 阿立哌唑与7种杂质分离试验色谱图Fig 2 HPLC chromatogram of aripiprazole and seven impurities
2.3.2 强制降解试验 取阿立哌唑约150 mg,精密称定,置50 mL量瓶中,加乙腈15 mL超声使溶解,加稀释剂定容至刻度,摇匀,作为供试品储备液。
① 未破坏试验:精密吸取上述供试品储备液1 mL置10 mL量瓶中,加乙腈3 mL,用稀释剂定容至刻度,摇匀,作为未破坏溶液。
② 热破坏试验:取在高温80℃条件下放置2 d的样品约15 mg,精密称定,按“2.2.1”项下方法处理,作为固体热破坏溶液。精密吸取供试品储备液1 mL置10 mL量瓶中,于80℃条件下放置24 h,冷却至室温,加入3 mL乙腈,再加稀释剂定容至刻度,摇匀,作为液体热破坏溶液。
③ 酸破坏试验:精密吸取供试品储备液1 mL置10 mL量瓶中,加1 mol·L-1盐酸1 mL,于80℃条件下放置24 h,冷却至室温,加1 mol·L-1氢氧化钠1 mL中和,加入3 mL乙腈,再加稀释剂定容至刻度,摇匀,作为酸破坏溶液。
④ 碱破坏试验:精密吸取供试品储备液1 mL置10 mL量瓶中,加1 mol·L-1氢氧化钠1 mL,于80℃条件下放置24 h,冷却至室温,加1 mol·L-1盐酸1 mL中和,加入3 mL乙腈,再加稀释剂定容至刻度,摇匀,作为碱破坏溶液。
⑤ 光破坏试验:取在(4500±500)Lx照度下放置约4 d并用紫外灯以辐照度90 uw/cm2辐照20 min的样品约15 mg,精密称定,置50 mL量瓶中,加入15 mL乙腈,超声使溶解,再加稀释剂溶解并定容至刻度,摇匀,作为固体光破坏溶液。精密吸取供试品储备液1 mL置10 mL量瓶中,于(4500±500)Lx照度下放置48 h并用紫外灯以辐照度90 uw/cm2辐照20 min,加入乙腈3 mL,再加稀释剂定容至刻度,摇匀,作为液体光破坏溶液。
⑥ 氧化破坏试验:精密吸取供试品储备液1 mL置10 mL量瓶中,加30%双氧水0.2 mL,80℃水浴10 min,加入3 mL乙腈,再加稀释剂定容至刻度,摇匀,作为氧化破坏溶液。
取稀释剂按上述破坏条件同法操作,作为各破坏试验的空白溶液。
取上述各溶液,照“2.1”项下色谱条件进样检测,结果见图3和表2,可知本品在光照和加热条件下稳定,无明显新增杂质产生,酸碱破坏下产生极少量杂质A和相对保留时间为0.37和0.43的未知杂质,在氧化破坏下不稳定,产生杂质F(5.04%),各破坏下产生的降解杂质与主成分峰均得到良好分离,主峰纯度合格,物料守恒。
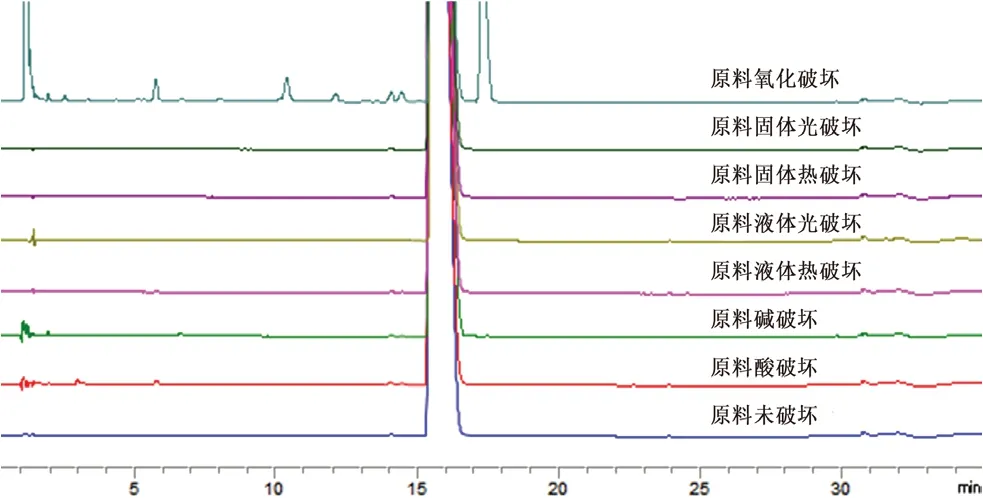
图3 阿立哌唑强制降解叠加色谱图Fig 3 Overlying HPLC of forced degradation test for aripiprazole

表2 强制降解试验结果Tab 2 Forced degradation test
2.4 线性关系考察
取阿立哌唑、杂质E、杂质F和杂质G对照品各约6 mg,精密称定,置100 mL量瓶中,加入30 mL乙腈,超声使溶解,加稀释剂定容至刻度,摇匀,得到各组分质量浓度约为60µg·mL-1的溶液,作为混合对照品储备液。精密吸取上述储备液适量,配制成一系列浓度水平的线性溶液,进样测定,记录色谱图。以质量浓度C对峰面积A作图,求得线性方程,计算回归系数(r)。
2.5 校正因子的测定
按“2.4”项下方法配制一系列浓度水平的线性溶液,分别采用两台不同厂家的液相色谱仪测定,按F=k主/k杂计算出各杂质的校正因子,结果见表3,可知各组分的r均大于0.998,杂质E、F和G的校正因子分别为1.33、1.06和0.90,故需采用加校正因子的主成分自身对照法计算杂质E的含量。

表3 特定杂质校正因子测定结果Tab 3 Correction factor for specific impurity
2.6 灵敏度试验
配制一定浓度的阿立哌唑和杂质A、B、C、D、E、F、G对照品溶液进样检查,分别以信噪比为3∶1时溶液的质量浓度为检测限;以信噪比为10∶1时溶液的质量浓度为定量限,结果阿立哌唑和杂质A~G的检测限分别为0.039、0.010、0.039、0.037、0.036、0.039、0039和0.037µg·mL-1,定量限分别为0.078、0.019、0.077、0.074、0.073、0.077、0.078和0.074 µg·mL-1,可满足灵敏度要求。
2.7 重复性和精密度试验
精密称取阿立哌唑约15 mg,置10 mL量瓶中,加乙腈3 mL使溶解,再加稀释剂定容至刻度,作为供试品储备液。精密吸取上述供试品储备液2 mL和“2.2.4”项下混合杂质对照品储备液1 mL,置10 mL量瓶中,加稀释剂定容至刻度,即得重复性试验用供试品溶液,平行配制6份。精密吸取上述供试品溶液0.5 mL,置50 mL量瓶中,加乙腈15 mL,再加稀释剂定容至刻度,即得自身对照溶液。按“2.1”项下色谱条件进样,结果6份平行样品杂质A~G的含量(约0.1%)的RSD分别为2.2%、2.3%、2.9%、2.4%、3.1%、2.7%和2.7%,总杂RSD为2.2%,符合规定。由另一位实验员于不同时间采用不同仪器进行有关物质的重复性试验,两位实验人员测定杂质A~G的含量平均值分别为0.14%、0.07%、0.09%、0.13%、0.13%、0.09%和0.10%,RSD分别为2.7%、2.6%、3.1%、3.1%、3.5%、4.5%和4.6%,总杂平均值为0.75%,RSD为3.1%,且放置过程中未产生大于报告限的新杂质,表明不同人员在不同时间采用不同仪器操作中间精密度良好。
2.8 溶液稳定性试验
取供试品溶液和自身对照溶液在不同时间进样,结果供试品溶液在室温条件下放置48 h内稳定性良好,杂质A~G的含量平均值分别为0.14%、0.07%、0.09%、0.13%、0.14%、0.09%和0.10%,RSD分别为0.5%、2.1%、1.4%、0.6%、4.6%、1.6%和2.4%;总杂平均值为0.77%,RSD为1.0%,且放置过程中未产生大于报告限(0.03%)的新杂质。
2.9 准确度试验
精密吸取“2.2”项下的混合对照品储备液5 mL,置50 mL量瓶,加稀释剂定容至刻度,作为回收率测定用混合对照品储备液。精密称取阿立哌唑(批号为AP20716001)约15 mg,置50 mL量瓶中,加稀释剂适量使溶解,平行配制9份,再分别加入回收率测定用混合对照品储备液2、2.5、3 mL,加稀释剂定容至刻度,配制成80%、100%和120%三个浓度供试品溶液,每个浓度平行配制3份,摇匀,作为供试品溶液。分别精密吸取上述9份供试品溶液0.5 mL置50 mL量瓶,加乙腈15 mL,再加稀释剂定容至刻度,即得对应的对照溶液。按“2.1”项下色谱条件进样,结果杂质E、F和G在80%浓度水平下的平均回收率分别为99.0%、94.0%和95.0%,RSD分别为2.2%、0.70%和5.1%;在100%浓度水平下的平均回收率分别为99.0%、95.0%和91.0%,RSD分别为1.7%、1.7%和0.50%;在120%浓度水平下的平均回收率分别为100.0%、96.0%和90.0%,RSD分别为1.1%、0.60%和0.30%均符合药典规定。
2.10 耐用性试验
为评估色谱条件的微小变动对测定结果的影响,分别考察不同品牌色谱柱、流速±0.1 mL·min-1、柱温变化±2℃、三氟乙酸含量变化±0.01%和梯度中有机相比例变化1%对阿立哌唑与已知杂质的分离度结果及样品检测结果的影响,取供试品溶液和对照溶液进样检测,结果阿立哌唑与杂质分离度均大于1.5,各条件下主要单个杂质和总杂的RSD均在0.7%~7.5%,并且不同条件下没有大于报告限的新杂质峰产生,表明此方法耐用性良好。
2.11 有关物质的检测
按“2.2”项下方法配制供试品溶液、自身对照溶液和空白溶液,照“2.1”项下色谱条件对阿立哌唑进行有关物质测定,并计算各杂质的含量,结果多批样品的单个杂质均小于0.10%,总杂质均小于0.50%,多批样品的有关物质均符合规定。具体结果见表4。

表4 有关物质测定结果(%)Tab 4 Determination of related impurities for aripiprazole (%)
3 讨论
3.1 检测波长的选择
分别取杂质A、B、C、D、E、F、G、阿立哌唑对照品溶液在二极管阵列检测器下检测,结果除杂质E以外,其余已知杂质和阿立哌唑均在254 nm波长附近有最大吸收。由于杂质E与主成分阿立哌唑响应值存在较大差异,因此本文采用254 nm作为检测波长,计算杂质E含量时需采用杂质外标法或加校正因子的主成分自身对照法。
3.2 色谱条件的选择
参考各国药典阿立哌唑原料和制剂的质量标准可知《中国药典》2015年版-阿立哌唑原料(口崩片、片、胶囊)和USP40阿立哌唑口崩片有关物质项下均采用枸橼酸盐-乙腈体系以等度洗脱的方式控制有关物质,USP40阿立哌唑口崩片有关物质项下采用0.05%三氟乙酸-乙腈体系以梯度洗脱的方式控制有关物质,而USP40和EP8.0阿立哌唑有关物质项下采用与USP40阿立哌唑口崩片有关物质项下相同的流动体系,并大大缩短分析时间,由60 min缩短为35 min。本文分别采用枸橼酸盐-乙腈体系和0.05%三氟乙酸-乙腈体系进行初步筛选,考察其对各已知杂质的分离情况,结果枸橼酸盐-乙腈体系杂质G在色谱柱上保留过强,40 min内还未出峰;采用0.05%三氟乙酸-乙腈体系,各杂质与主成分均分离良好,且阿立哌唑峰形较好,故初步确定流动体系为0.05%三氟乙酸-乙腈。本研究在此基础上提高供试品溶液浓度使灵敏度达到要求而避免杂质漏检,并进一步优化梯度洗脱程序使阿立哌唑和杂质F达到基线分离。
3.3 校正因子的研究
本研究首次采用不同品牌的仪器(Agilent和Waters)通过斜率法建立阿立哌唑各特定杂质的校正因子,达到试验更简便快捷并降低检验成本的目的,结果杂质F和G的校正因子在0.9~1.1,杂质E的校正因子为1.33,超出了0.9~1.1的范围,故采用加校正因子的主成分自身对照法计算杂质E的含量。
3.4 杂质分析
根据阿立哌唑的合成工艺,可知阿立哌唑中可能存在的工艺杂质为A~G,根据强制降解试验结果可知杂质A、E、F同时也是降解杂质,在碱破坏条件下产生杂质A,酸破坏下杂质E增大及氧化破坏下极易产生杂质F,提示本品应该密封保存。
多批样品中均只检测出已知杂质E和杂质G,相对保留时间为1.23的未知最大单杂,且以上各杂质含量均小于0.10%。杂质E为阿立哌唑脱氢化物,在酸性条件下容易生成,提示在阿立哌唑制剂的生产过程中应特别注意pH环境,避免pH过低。
综上所述,本研究建立了一种有效监测阿立哌唑中有关物质的RP-HPLC法,并对该方法的有效性和检出能力进行了全面的评价。结果该方法准确度高、专属性强、耐用性好,可以同时控制阿立哌唑中的7个杂质,为阿立哌唑的质量控制提供参考。
