镁基水滑石催化材料的研究进展
2021-06-30高娃冉祥堃赵汗青赵宇飞
高娃,冉祥堃,赵汗青,赵宇飞
(1北京农学院生物与资源环境学院,北京102206;2北京化工大学化工资源有效利用国家重点实验室,北京100029)
引 言
我国是世界上盐湖最多的国家,盐湖资源丰富,氯化钾、氯化镁、氯化锂探明资源储量分别占全国的50%、90%和50%,潜在经济价值数十万亿元。盐湖资源的科学开发与利用,不仅影响到我国新能源、新材料的发展和应用,而且作为国家战略资源,关系到我国未来综合国力的提升。长期以来,盐湖资源开发都以钾肥生产为主,然而每生产1吨钾肥就要产生12吨副产物水氯镁石。受工艺技术限制,这部分镁资源未实现高效开发利用,大量的氯化镁会被重新排放到盐湖中,形成“镁害”。只有当盐湖镁产业在规模上发展到与盐湖钾肥等产业相匹配时,盐湖资源才能真正走上循环经济发展模式。因此,合理利用镁资源制备高性能、高附加值的功能性材料,是镁资源的可持续性开发与利用的重要导向标。
水滑石(layered double hydroxides,LDHs)是一类二维阴离子层状材料,化学组成可表示为M2+1-xM3+x(OH)2]x+[An-]x/n•mH2O。其主体层板与水镁石Mg(OH)2结构相似,由羟基配位八面体基元构成,该八面体的M2+部分被M3+取代,从而使层板带正电荷,常见的M2+和M3+有Mg2+、Cu2+、Co2+、Ni2+、Al3+、Fe3+等。An-是层间阴离子,常用的有OH-、CO2-3、NO-3、Cl-、SO2-4等无机离子。带负电荷的阴离子可与层板的正电荷相平衡,使水滑石整体显电中性。镁基水滑石指层板结构包含Mg2+的二元、三元或多元水滑石,其具有层板金属元素原子级分散、层板组成比例可调、插层阴离子可交换以及结构记忆效应等结构特点,由此可构筑多种镁基功能材料[1-3]。近年来,随着纳米材料以及二维层状材料的飞速发展,镁基水滑石由于自身结构的独特性,在现代石油化工、煤化工、精细化工以及环境净化等诸多催化领域表现出了广泛的应用前景[1-3]。
本文综述了镁基水滑石催化材料近年来的研究进展(图1):(1)镁基水滑石直接作为催化剂应用于酸碱催化和超分子插层手性催化,论述了层板元素组成、层间阴离子类型对镁基水滑石催化材料酸碱性和催化活性的影响规律;(2)焙烧后得到的复合金属氧化物(mixed metal oxides,MMOs)应用于固体碱催化和环境净化催化领域,系统论述了层板元素组成、比例、焙烧温度等条件对催化剂结构的影响,阐明了催化剂的结构特性(比表面积、表面元素价态、酸碱性、氧缺陷等)与催化活性的关联;(3)将MMOs作为载体制备负载型金属催化剂,总结了MMOs作为载体的优势及其对催化剂活性位结构和催化性能的影响;(4)镁基水滑石复合材料,分析了镁基水滑石与纳米碳材料、金属硫化物和磁性材料等复合后所展现出的协同催化效应。希望本综述能够吸引更多研究者关注镁基水滑石催化材料,鼓励人们在盐湖镁资源利用领域开展更多富有创新性的研究。
1 镁基水滑石催化剂
1.1 镁基水滑石的结构特点
镁基水滑石具有独特的结构特点(图1):(1)层板金属元素种类和比例可调,根据水滑石层板构筑原则,可以将部分Mg2+替换为Cu2+、Ni2+、Co2+等其他二价金属离子。近年来报道中提到贵金属Rh、Pd、Pt等也可少量进入水滑石层板。不同金属元素催化性质不同,使镁基水滑石材料广泛应用于各类催化反应中,如引入Cu、Fe和Co元素后是优异的合成气转化催化剂[4-6],引入Pd元素后具有不饱和烃加氢性能[7-9]。此外,由于主体层板表面有丰富的羟基,单纯的二元镁基水滑石材料MgAl-LDHs即可用作有效的固体碱催化剂,催化多种有机反应如Knoevenagel缩合、迈克尔加成、酯交换反应等[10-13]。(2)镁基水滑石的层间具有极大的可调空间,可以插入无机阴离子、有机酸或碱、杂多或同多酸离子和生物分子等。以镁基水滑石材料作为前体,经插层组装制备超分子插层结构催化材料,是目前相关领域的研究热点之一。(3)层板金属离子在晶格定位效应的作用下呈现原子水平上均匀排布、高分散的状态。将镁基水滑石前体在不同气氛(空气、氢气或氮气)和不同温度下焙烧,经结构拓扑转变过程,可以获得高分散、高稳定的MMOs或负载型金属纳米催化材料。MMOs具有较大的表面积和丰富的Lewis碱性位点[11,14-16]。若将MMOs置于无CO2的水溶液中会发生水合复原,恢复到LDHs结构,在这个过程中其层间引入了大量OH-阴离子,从而提供丰富Brønsted碱性位点。因此,Mg基LDHs、MMOs以及通过复原过程活化的LDHs材料,在多相催化领域的应用得到了众多国内外研究者们的广泛关注。

图1 Mg基水滑石催化材料结构特点及应用示意图Fig.1 Propertiesand applications of catalysts fabricated by using Mg-LDHs as supports/precursors
1.2 镁基水滑石催化剂的应用
Mg基LDHs直接作为催化剂时,主要应用于固体酸碱催化反应和超分子插层手性催化反应。Mg基LDHs的主体层板一般由镁氧八面体和铝氧八面体组成,且主层板之间存在着氢氧根离子来中和层板所带的正电荷,使Mg基LDHs本身呈弱碱性和Lewis酸性。其中,碱性强弱与层板中二价金属氧化物的碱性强弱基本一致;酸性强弱则同时与层板中三价金属氢氧化物的酸性有关。此外,利用Mg基LDHs层间阴离子的可交换性,可将不同类型的无机阴离子、有机阴离子、同多或杂多酸阴离子等插入LDHs层间,得到同时具备插层阴离子特点和LDHs层板结构特点的双功能催化材料。通过调变层间阴离子的体积、数量、价态以及阴离子与层板羟基之间的键合强度,使其在手性催化反应中展现出了优异的催化性能。
1.2.1 酸碱协同催化材料 绿色可持续发展是催化领域的永恒课题,与液体碱相比,固体碱催化剂具有对环境友好、腐蚀性小、易于回收等优势。Mg基LDHs的主体层板具有丰富的羟基,可用作有效的固体碱催化剂。通过调变层板金属离子和层间阴离子的类型、比例,可以调控LDHs表面酸碱性质,进而影响其催化性能。二元Mg基LDHs可以通过引入第三种元素来对其主体层板结构进行调控。近期,Wang等[17]报道了使用稀土元素La、Y、Ce等元素对MgAl-LDHs进行改性之后再进行焙烧及水合复原,考察了其对丙酮自缩合反应的催化性能的影响。结果表明,引入少量的La、Y和Ce元素可以增强中等强度碱性位点的数量与强度,并且使MgAl-LDHs主体层板的混乱度增加,暴露更多位于层板的边缘或角落的低配位氧原子,提供更强的Lewis碱性位点,进而提升了其催化性能。郭军等[18]以异丙醇脱水反应为探针反应研究了不同类型层间阴离子对LDHs酸碱性的影响,结果表明SO2-4、NO-3、Cl-型LDHs以酸中心为主,且酸性大小顺序为SO2-4>Cl->NO-3,而CO2-3型LDHs则以碱性为主。层间阴离子的共轭酸性越强时,对产物丙烯的选择性越高。目前,Mg基LDHs直接用于酸碱协同催化反应的研究很少,未来还有很大的探索空间。
近年来,研究发现LDHs的层间既可以存在小尺寸无机阴离子也可以容纳大尺寸的分子,如多金属氧酸盐(多酸)分子,进而构筑酸碱协同催化剂。多酸是一大类由钒、钼、钨和铌等组成的阴离子型金属氧化物,是优异的氧化催化剂,常用于有机化合物的部分氧化反应。但是多酸本身具有的酸性易使其对产物深度反应,产物选择性很差。将多酸与水滑石插层组装构筑多酸基酸碱协同催化剂可以有效解决该问题。Li等[19-20]设计合成了多酸水滑石插层组装催化剂Mg0.73Al0.27(OH)2[WZn(ZnW9O34)2]0.023·1.1H2O,研究发现进入MgAl-LDHs层间的多酸端基O原子以氢键的形式与LDHs层板的羟基结合,限制了质子在多酸表面的移动,从而降低了多酸本身的酸性,抑制了产物深度反应,进而提高了其对相应环氧化物的选择性(图2)。尽管多酸本身的酸碱减弱了,但是却保持了原有的高活性,实现了酸碱协同催化的目的。这是单独LDHs载体或者LDHs载体与多酸的物理混合物都无法达到的催化效果。

图2 杂多酸插层水滑材料作为烯丙醇环氧化酸碱协同催化剂[20]Fig.2 The as-synthesized polyoxometalate intercalated LDHs nanomaterials asbase-synergistic catalysts toward the epoxidation of allylic alcohols[20]
Zhao等[21]进一步拓展了多酸水滑石插层组装催化剂的种类和应用,将多种Finke型多酸插入到MgAl-LDHs层间。将其作为多酸基酸碱协同催化剂,一方面可以在温和条件下高选择性地催化醛酮和原位生成羟胺的肟化反应(图3)。另一方面,MgAl-LDHs主体层板与多酸客体分子之间以及客体与客体分子之间存在多重相互作用力,多酸分子不易流失,极大地提升了催化剂的稳定性,可以重复使用10次而催化活性无明显降低。该课题组还将Weakly型多酸引入到不同碳链长度柔性离子液体修饰的MgAl-LDH层间,构筑了新型稀土多酸插层水滑石固体碱催化剂Mg3Al-ILs-Cn-Law10(n=4,8,12)。将其应用于烯醇环氧化反应中,实现了在温和、无溶剂条件下高效催化烯醇环氧化[22-24]。Bai等[25]将十二磷酸阴离子簇(POMs)交换到MgAl-LDHs层间,该插层材料不仅将POMs成功多相化,而且在H2O2氧化环己醇反应中展现出了较高的催化活性。在此基础上,用Ni2+替代层板上的部分Mg2+(NiMg2Al-POM LDH)来降低LDH层板的碱度,削弱层板与POM阴离子的酸碱反应,进一步提高了催化剂的催化活性和结构稳定性。多酸/Mg基LDHs纳米复合材料目前已广泛应用于烯烃环氧化、硫醚氧化、酸催化、脱氢反应等,并显示出了优异的催化效果。围绕多酸/Mg基LDHs催化剂的后续研究,进一步深入研究其结构和电子转移特性,尤其是多酸客体分子具体排列取向、分布特点,扩展多酸/Mg基LDHs复合材料在不同领域的功能应用是研究重点。

图3 杂多酸插层MgAl-LDHs在芳香醛肟化反应中展现出的协同催化效应示意图[21]Fig.3 Sandwich-type polyoxometalates intercalated in layered double hydroxides have been shown to exhibit synergistic catalysis of the oximation of aromatic aldehydes with a large enhancement of selectivity[21]
1.2.2 超分子插层手性催化材料 均相催化剂多相化是近年来不对称催化领域的研究热点,但其面临的问题是多相化往往会造成催化活性大幅度下降,对映选择性降低甚至完全丧失。基于Mg基LDHs层间阴离子种类和数量的可调控性,在LDHs层间引入具有不对称催化活性中心阴离子,利用层内空间的限域效应和主体层板的协同效应可以极大地提高反应活性和选择性。Pitchumani等[26]利用焙烧复原法将16种L-氨基酸阴离子插入MgAl-LDHs层间,应用于催化苯酚及苯硫酚的选择性甲基化反应,均获得了远高于游离氨基酸催化的反应转化率和选择性。其中亮氨酸插层LDHs催化反应主产物产率高达98%,远高于均相亮氨酸3%的反应产率。研究表明,氨基酸阴离子疏水链越长反应产率越高,层内限域空间的氨基酸活性中心疏水环境增强可提高反应活性和选择性。He等[27-28]的研究成果进一步说明LDHs层内空间具有明显的空间限域效应,将手性酒石酸钛配合物通过静电作用引入MgAl-LDHs层间,酒石酸钛阴离子在LDHs层间保持了其原有的配位结构,且在LDHs层间实现了双层交错的有序排列。将其应用到苯基甲基硫醚的选择性氧化反应中,反应的不对称选择性从均相的几乎为0增大到了50%。LDHs主体层板的空间/电子协同效应可以进一步强化不对称选择性,该课题组提出以具有二维有序刚性层板的LDHs作为配体大取代基,可使活性中心周围空间环境存在稳定的差异,是获得高对映体过量百分数(ee值)产物的前提。层间客体与主体层板之间弱相互作用的能量差与两种对映异构体过渡态之间的能量差相近,进而影响了对映体选择性[29]。随后,他们进一步利用LDHs的空间效应/碱性协同催化的思路,将氨基酸改性的LDHs层板作为Rh中心配体催化C—H键活化反应(图4),[Cp*RhCl2]2为二氯(五甲基环戊二烯基)合铑(Ⅲ)二聚体,Piv代表N-(pivaloyloxy)benzamide。LDHs层板除了提供大的空间位阻提高了不对称选择性(rr>20∶1),同时提供了碱性位提高反应活性(yield>99%)。通过调变LDHs层板二价金属种类(Zn2+、Mg2+、Ni2+)可有效调控层板碱性,反应活性随LDHs碱性的增加而增加。结果表明,水滑石作为固体碱,能够提供反应所需的碱,不需要额外加碱就可以获得了比液体碱更高的活性[30]。

图4 氨基酸改性的MgAl-LDHs层板作为Rh中心配体催化C—H键活化反应的示意图[30]Fig.4 Rhodium(Ⅲ)-catalyzed C—H activation has been promoted impressively with ligand-attached nanosheets of LDHs,a natural and/or synthetic anionic layered compound[30]
超分子插层手性催化材料利用Mg基LDHs层内空间的限域效应和主体层板的协同效应强化手性活性中心的不对称诱导性,利用多相反应体系类均相化和LDHs层板功能协同提高反应活性,而且解决了传统的均相催化剂在使用过程中出现的催化剂失活、反应后难于回收再利用,以及反应体系分离过程耗费大量溶剂等问题,符合绿色化学发展趋势的要求。
2 镁基复合金属氧化物催化剂
将Mg基LDHs前体在450~600℃温度范围内焙烧可以得到均匀分散的MMOs,其具有较大的表面积和丰富的Lewis碱性位点。与LDHs前体的酸碱活性位结构不同,MMOs中含有三种强度的碱性位:OH-提供的弱碱性位,Mg-O离子对提供的中强碱性位,离子提供的强碱性位[12]。进一步,利用水滑石的“记忆效应”,将MMOs置于去除CO2的水溶液中,MMOs可重新转化为LDHs结构,水合复原过程中在LDHs层间引入了大量OH-阴离子,提供了丰富Brønsted碱性位点。通过对水合复原过程条件的调变,可以对Brønsted碱性位点的数量和强度进行有效调控。焙烧或焙烧复原后得到的固体碱催化剂在羟醛缩合反应、迈克尔加成反应、酯交换反应、氧化反应、甲烷二氧化碳重整反应和CO2催化转化等反应中均展现出了优异的催化性能[10-13]。固体碱催化剂的催化性能与其碱性位结构、强度及碱性位数量等密切相关。合理设计的固体碱催化剂的碱性位点结构,可以有效地提升其碱催化性能,特别是提高活性和选择性[14-16,31]。
2.1 固体碱催化材料
如何有效调控碱性活性位的结构,进而强化催化剂的反应性能和稳定性,同时深入认识碱性位结构调控规律及催化作用机制是制备高性能Mg基LDHs固体碱催化剂的研究重点。通过调变Mg/Al比例来调控MMOs的碱性位结构是最常用的调控手段之一[14,32]。黄艳丽等[33]制备了一系列不同n(Mg)/n(Al)比的Mg基LDHs前体,焙烧后得到了Mg(Ni,Al)O复合氧化物催化剂,用于甲烷二氧化碳重整反应中。结果表明,随着Mg/Al比例的增加,MgAl2O4相转变为Ni(Mg,Al)O固溶体,导致金属与载体间作用力增强、催化剂碱性位点数增多、晶粒尺寸逐渐减小,进而使催化活性升高和抗积炭能力加强。而当n(Mg)/n(Al)=1时,催化剂的活性和稳定性最佳,且催化剂表面生成的积炭量最少。Zheng等[32]研究发现在以NiMgAl-LDHs为前体制备的Ni4MgAlO催化剂中(图5),随着Mg/Al比例的增加,乙醇的转化率降低,但是高碳醇的选择性会随之升高。当Mg/Al比为4时,乙醇转化率为18.7%,对正丁醇和C4~C8高级醇的选择性分别达到了55.2%和85%。采用微量吸附量热技术,以CO2和NH3为探针分子定量地表征了样品表面酸碱中心的强度和数量,并与其催化性能进行了关联。结果表明,不含Mg的NiAlO样品显示中等强度酸性和强碱性位,比例为1∶2.1;而Ni4MgO样品有强碱性位,没有酸性位;只有Ni4MgAlO催化剂有适中的酸性位和强碱性位,比例为1∶3.8,两者协同催化乙醇转化,抑制了乙醛缩合物的生成,提高了高级醇的选择性[32]。

图5 Ni4MgAlO催化剂酸碱协同催化转化乙醇到高级醇反应的示意图(a)及Ni含量和催化剂酸碱性位对催化性能的影响(b)[32]Fig.5 Upgrading ethanol to n-butanol over highly dispersed Ni4MgAlOcatalysts(a);The effects of Nipromotion and acid–base site distributions of catalysts were investigated using various techniques(b)[32]
除了调变MgAl-LDHs的Mg/Al比例外,通过在层板引入其他过渡金属元素或稀土元素来改变催化剂表面晶格氧浓度,或使催化剂无序度增加,暴露出更多的低配位氧原子,进而可以提供更多的碱性位,是另一个调变催化剂碱性位的有效手段。肖福魁等[34]制备了一系列不同价态过渡金属(Fe、Cu、Zr)改性的Mg-Al固体碱催化剂,并用于酯交换法合成碳酸二甲酯的反应。CO2-TPD表征结果表明过渡金属的加入虽然使总的碱量有所下降,但单位比表面积上催化剂的碱含量明显提高,催化剂上总的碱密度顺序为:FeMgAl>CuMgAl>ZrMgAl>MgAl。进一步分析XPS中O 1s谱图发现,Fe、Cu、Zr作为电子助剂掺入Mg-Al氧化物中,提高了单位面积上晶格氧的浓度。同时还影响了Mg-Al氧化物中晶格氧的电负性,使O原子周围电子密度增加。催化剂供电子能力越强,其碱性越强,表面碱密度越高,酯交换法合成碳酸二甲酯催化活性就会增强。催化活性与总碱密度,特别是中、强碱密度之和呈正相关关系。Wang等[35-36]考察了过渡金属(Fe3+、Cr3+、Cu2+、Co2+、Ni2+、Mn2+、Zn2+)对Mg-Al复合氧化物碱性位强度和数量的影响规律,发现MgAl-Zr复合金属氧化物催化剂中、强碱性位数目之和最高,在氨基甲酸甲酯酯交换合成碳酸二乙酯中表现出了最好的活性(图6)。近期,也有文献报道,通过引入镓、铟、KF等均可以使水滑石固体碱催化剂展现出更多碱性位点[37-38]。

图6 Ni4MgAlO催化剂酸碱协同催化转化乙醇到高级醇反应的示意图[36]Fig.6 Schematic illustration of the synthesis of mesoporous Mg-Al-Zr mixed oxides with tunable acidic-basic property[36]
众所周知,当催化材料的尺寸或厚度减小时,表面会暴露更多的催化活性位点,表面原子也很容易脱离晶格而形成空位/缺陷。空位/缺陷的形成会改变晶格氧的电子结构,进而影响碱性位点的活性,实现对催化性能的调控。Lei等[39]采用成核/晶化隔离法(SNAS)和尿素分解法两种方法合成了MgAl-LDHs前体,再对其进行焙烧及水合复原的活化过程。SNAS是由Duan等[40]研发,因在成核过程中反应物瞬时充分碰撞、接触,所以会产生大量的晶核,这保证了悬浮液在晶化过程中晶体尺寸的均一[39]。在制备脂肪酸单乙醇酰胺反应中,用SNAS方法合成的LDHs晶粒尺寸更小更均一(图7),比表面积更大,其表面暴露的碱性位点也更多,在120℃的反应温度下,催化反应物转化率可达87%。将LDHs剥离成为单层纳米片时,可以使催化剂表面活性位点的暴露达到最高程度,Ruiz等[41]将剥离后的单层MgAl-LDHs焙烧得到MgAl-MMO固体碱催化剂,剥离后的催化剂表面具有丰富的Lewis碱性位点,使其在苯甲醛与2-丙醇的Meerwein–Ponndorf–Verley(MPV)还原反应中展现出优异的催化活性。

图7 成核/晶化隔离法和尿素分解法制备的MgAl-LDHs前体[(a)、(b)]及相应的焙烧复原后LDHs[(c)、(d)]的扫描电镜照片[40]Fig.7 SEM images of S-MgAl-LDH(a),U-MgAl-LDH(b),S-MgAl-RLDH(c),and U-MgAl-RLDH(d)[40]
因此,通过调变Mg基LDHs材料中层板金属离子种类、组成比例、前体制备方法和焙烧温度等来调控MMOs碱性位结构的性质、强度和相对数量是非常有效的手段。但已有文献报道中对MMOs碱性位结构的认识及催化作用机制的研究还不够深入。采用多种原位表征技术深入研究LDHs固体碱催化剂本征活性位的精细结构,揭示其配位结构、电子密度对碱性位密度和强度的影响规律,进而阐明碱性位催化作用机制是未来的研究重点。
2.2 环境净化材料
近年来,传统化石燃料的大规模利用,导致能源短缺、环境污染等诸多问题。水体重金属污染、大气污染、温室气体增加,严重危害着生态环境以及人类的健康。LDHs及其焙烧产物具有较大的比表面积、丰富的孔径分布、高热稳定性、良好的金属氧化物分散性等结构特点,因此其在水体环境污染、挥发性有机化合物(VOCs)、温室气体CO2的治理领域具有重要的应用。作为吸附剂在水体环境污染治理方面,可对重金属污染物(Hg、Pb、Cd、Cr等)、有机污染物(甲基橙、甲基蓝、罗丹明B等)、无机阴离子(磷酸盐、卤素、硫氰酸盐、砷酸盐等)和放射性元素[U(Ⅵ)、I]等多种污染物进行吸附。在吸附过程中,主要通过电子相互作用、氢键和络合作用吸附污染物。吸附阴离子污染物过程中,目标污染物可将层间阴离子置换出来使污染物得以去除。尤其,LDHs在300~500℃条件下焙烧后转变为层状双金属氧化物(LDO),当其与水或含有适当阴离子的溶液接触时,又可以重构层状结构,称为LDHs的“记忆效应”。利用这一性质,当目标污染物与LDOs相互作用时,可以插入LDOs塌陷的层板之间,在重构结构的同时实现了对污染物的去除(图8)[42]。张曼等[43]制备的焙烧态CaMgAl-LDO对含砷废水中As(Ⅴ)的最大吸附量能达到71.86 mg/g。研究吸附机理发现,溶液中带负电的砷酸氢根离子插入CaMgAl-LDO层间,利用LDHs的结构“记忆效应”特点使其层状结构重建,在该过程中实现了对As(Ⅴ)的吸附。对焙烧后LDO进行适当的修饰,例如层板修饰、层间插层等,可以进一步提升其对有机、无机污染物的吸附量。Yan等[44-46]将乙二胺四乙酸(EDTA)插层到MgAl-LDH中构筑了有效去除水中重金属的高性能纳米材料。带负电荷的铬酸根与质子化的LDHs表面之间通过静电吸引发生络合反应,随后铬酸根与EDTA2–发生层间阴离子交换,EDTA的—COOH提供的电子供体使部分剧毒的Cr(Ⅵ)还原成环境友好的Cr(Ⅲ)[44-46]。Ma等[47-49]制备了层间阴离子为MoS2-4的MgAl-LDHs,对重金属阳离子(Pb2+、Hg2+、Cd2+、Ag+等)及金属含氧酸根离子(HAsO2-3/HAsO2-4、CrO2-4)都具有较好的选择吸附性能,既能实现对目标重金属的选择性吸附又能避免被氧化,从而实现目标离子的高效脱除。

图8 LDHs记忆效应插层阴离子B(OH)4-、H3BO3对硼的去除机理示意图[42]Fig.8 Removal mechanismof boron speciesby different CLDHs dependent on the divalent metals and initial pH of boron solution[42]
挥发性有机化合物(VOCs)是光化学烟雾、对流层臭氧以及二次气溶胶的前体,经过复杂的物理化学转换过程会导致二次污染,部分挥发性有机化合物具有温室效应,会加剧全球变暖。此外,挥发性有机化合物具有毒性、致癌性和致突变性等特点,对人体和环境均会产生严重的影响。因此,挥发性有机化合物的治理已经成为世界范围内的研究热点,将VOCs非均相催化氧化成CO2和H2O,是一种最为有效的分解VOCs的方法。Mg基LDHs基于其大表面积和多孔性、高热稳定性、良好的金属氧化物分散性,在VOCs催化分解领域有非常大的潜力。近期,Shuan等[50]综述了四大类LDHs衍生的VOCs降解催化剂,含过渡金属的LDHs焙烧转变成MMO催化剂、贵金属/MMO催化剂、核壳型MMO催化剂,以及整体型MMO催化剂。贵金属催化剂活性高,但价格昂贵。因此,通过对含过渡金属的LDHs的层板元素组成、层板元素比例、焙烧温度、制备方法等条件的调变,来实现对催化剂的比表面积、孔结构、表面元素价态、氧缺陷、还原性等性质的调控,进而获得高活性、低成本的催化剂是未来研究趋势。例如,Genty等[51]研究发现,通过选取不同的二价金属离子,如Mn2+、Cu2+、Co2+、Ni2+,来替代MgAl-LDHs中的Mg2+离子可以显著影响对VOCs的氧化性能。不同金属离子替代的MgAl-LDHs焙烧后的混合金属氧化物的催化性能明显优于单金属氧化物的性能,例如将Cu/Mn/Mg/Al和Co/Mn/Mg/Al-LDHs前体焙烧后形成的包含Cu-Mn和Co-Mn的混合金属氧化物的理化性质(比表面积、孔结构、表面元素价态、氧缺陷、还原性)得到了优化,其中Co/Mn/Mg/Al催化剂具有最佳的VOCs降解性能[52]。催化剂的制备方法对MgAl-LDHs催化分解VOCs的性能影响非常大,常见的制备方法有共沉淀法、微波辅助法和超声波辅助法。其中,微波辅助法制备的LDHs催化剂活性最高,由于其具有更大的比表面积和更小的晶粒尺寸。而对水滑石前体的焙烧温度主要影响LDHs衍生催化剂的结晶度、还原性、比表面积和氧空位浓度,从而影响VOC氧化活性。Palacio等[53]采用共沉淀法制备了MnCuAl-LDHs前体,并在450℃和600℃下进行了焙烧效果评价。450℃下焙烧的MnCuAl样品比600℃下焙烧的样品具有低结晶度的尖晶石相。经450℃处理后,MnCuAl样品具有最好的织构和还原性,在297℃时甲苯转化率达到100%,催化性能最高。
二氧化碳(CO2)是典型的温室气体,也是重要的储量丰富且可再生的碳资源,可用于生产酯、醇、醚、酸等重要的化工原料或液体燃料。实现CO2的资源化利用是国际前沿和研究热点。CO2捕集是目前最经济可行的技术,捕集的CO2通过后续转化利用合成重要化合物或提高原油采收率。传统MgO吸附材料由于含量丰富、价格低、理论捕集能力高(每克吸附剂可捕获1100 mg CO2)、无毒且操作温度范围宽(从室温到中间温度)而具有很大潜力。但是MgO吸附剂自身碱性活性位点比较少,且具有较高的晶格焓,造成CO2捕集能力低且吸附动力学差。Mg基LDHs具有吸附速率快、较低的再生能量、多次循环后的容量保持能力以及合适的CO2吸附动力学等特点受到广泛关注,已经发展成为一种较好的CO2捕集材料。Lara-García等[54]结合共沉淀法和浸渍法合成了Li-Na-K掺杂的MgAl-LDO作为高压CO2捕获材料,系统研究了不同压力和不同吸附温度对CO2捕集能量的影响。该材料在中等温度段展现出了高的CO2吸附量(7~8 mmol·g-1)和较好的稳定性(10次循环以后性能没有改变)。原位DRIFT表征结果表明,(Li-Na-K)NO3的添加起到了关键作用,其与NO-3相互作用形成NO-2和CO2-3,硝酸盐向亚硝酸盐转变时释放出O-。随后,CO2-3与MgAl-LDO作用形成MgCO3,O-物种再与亚硝酸盐结合形成初始状态的NO-3。该课题组还通过调控水热合成反应条件以类水滑石Mg5(CO3)4(OH)2·4H2O为前体制备了MgO基中温CO2吸附材料,系统研究了不同合成条件对MgO基吸附材料对CO2吸附容量和吸脱附循环稳定性的影响规律。结果表明,前体的焙烧温度影响了MgO吸附材料的缺陷位的产生,CO2-TPD数据表明该MgO吸附材料具有丰富的不同强度的碱性位。CO2被吸附后主要形成三种碳酸盐物种,包括碳酸氢盐、双齿和单齿的碳酸盐。最低焙烧温度(350℃)的MgO样品展现出高比表面积(372.0 m2·g-1),较大的孔径(0.38 cm3·g-1),丰富的碱性位,使该材料在非常宽泛的温度范围内(60~300℃),对于CO2的捕集量达到了1.22~1.99 mmol·g-1,是传统MgO吸附材料的20倍[55]。
以上工作表明,Mg基LDHs吸附材料的合成工艺简单、周期短、条件易于控制,在重金属污染物的吸附、挥发性有机化合物的催化氧化和CO2的捕集利用等方面展现出了优异的性能,有着比较广阔的应用前景。在后续的研究工作中,制备高性能的LDHs复合材料,充分发挥LDHs和其他纳米材料的协同作用,进而优化Mg基LDHs吸附材料的吸附性能;采用多种谱学表征技术和理论计算等手段对吸附机理开展进一步的深入研究,为高性能吸附材料提供理论依据是研究重点。
3 镁基复合金属氧化物作为催化剂载体
负载型催化剂作为一类重要工业催化剂,广泛应用于合成氨工业、能源环境化工和精细化工等重要的工业生产过程。传统的Al2O3、MgO载体存在活性组分在载体上分散不均匀、金属和载体之间的弱相互作用使得金属在高温焙烧和反应过程中易发生团聚等问题。目前,以Mg基LDHs经结构拓扑转变得到的复合金属氧化物(MMO)作为催化剂载体,或将活性组分金属引入Mg基水滑石层板,直接制备高性能的负载型单金属或双金属催化剂,在氧化脱氢、选择性加氢和合成气转化等反应中展现出了独特的优势。该类催化材料具有金属纳米颗粒粒径、形貌、几何/电子结构、载体酸碱性、金属与载体间相互作用可控的特点,为催化反应提供了所需的特定结构和活性中心。
3.1 负载型非贵金属催化剂
LDHs中的金属离子在主体层板中呈现原子级均匀分散,有利于在焙烧还原过程中形成稳定性高且分散性好的负载型金属催化剂。此外,活性金属与氧化物载体之间存在强相互作用,可以有效地抑制纳米金属颗粒的团聚和烧结。根据LDHs层板构筑规则,Fe、Co、Ni等活性非贵金属元素与LDHs材料具有非常好的兼容性,可以将其直接引入Mg基LDHs层板,经焙烧还原过程得到负载型催化剂[1,3]。
Li等[56]以CuMgAl-LDHs为前体合成了Cu纳米颗粒均匀、高度分散的负载型Cu/MgO-Al2O3催化剂(图9),其对1,4-环己烷二甲酸二甲酯(DMCD)加氢生成1,4-环己烷二甲醇(CHDM)反应展现出了优异的催化性能,CHDM的产率达到99.8%以上,并且保持200 h以上的催化剂稳定性,明显优于传统浸渍法制备的负载型Cu催化剂。研究发现高分散的Cu0位点与MgO载体的Lewis碱性位点的协同催化效应是关键,其中Cu0位点是DMCD加氢的主要活性位,而MgO载体表面Lewis碱性位点很容易与羰基C O π*空轨道发生相互作用,在氧原子的亲核进攻下,形成极化的四面体羰基中间体,增加DMCD分子中酯基加氢能力和CHDM的生成选择性[56]。类似地,Xia等[57-58]以CuMgAl-LDHs为前体制备的Cu基金属催化剂,对甘油无氢合成1,2-丙二醇表现出较高的活性和稳定性。进一步证实,对于能够进入LDHs层板的Fe、Co、Ni、Cu等元素,经LDHs前体路径是制备高分散、高稳定的负载型金属催化剂的有效途径。Zhao等[59]以MoO2-4插层的FeMgAl-LDHs为前体,制备了高分散(1014~1016 m-2)、粒径大小可控(3~20 nm)、高稳定性的负载型Fe纳米催化材料(图10)。研究表明,除了LDHs层板的晶格限域作用外,插层客体MoO2-4与层板的主客体相互作用也有效抑制了Fe在还原过程中的烧结,进一步提高了其分散性和稳定性;该催化剂在催化生长双螺旋单壁碳管的反应中表现出优异的催化性能。由此可见,以LDHs为前体制备高分散、高稳定的负载型金属催化剂具有显著的优势。

图9 Cu/MgO-Al2O3催化剂协同催化DMCD加氢生成CHDM的机理示意图[56]Fig.9 Schematic representation of the hydrogenation mechanismover Cu-based catalysts derived from CuMgAl–LDH precursors[56]

图10 MoO42-插层FeMgAl-LDHs为前体制备高分散Fe纳米颗粒催化剂示意图[59]Fig.10 Schematic illustration to show the formation of high density Fe NPs by the incorporation of MoO42-in MgAlFe–LDH[59]
相比于负载型单金属催化剂,双金属催化剂具有可调控的几何结构、电子结构和界面结构,双金属间的协同催化作用可显著提升金属催化剂的性能。若将两种具有催化活性的过渡金属元素同时引入Mg基LDHs层板,则可以获得MMO负载的双金属催化剂。Wang等[60]以CoMgFe-LDHs为前体,在氢气气氛下还原得到了CoFe/MgO合金样品,而且通过LDHs前体中Co/Fe的比例调节实现了Co/Fe合金比例的调控。Gao等[61]以NiMgFe-LDHs为前体制备了NiFe/MgO合金催化剂,其在室温条件下实现了快速完全分解水合肼制氢气。高分辨透射电子显微镜(HRTEM)及高角环形暗场扫描透射电子显微镜(HAADF-STEM)数据显示NiFe合金纳米颗粒尺寸均一,MgO载体对NiFe合金颗粒起到了非常好的分散和稳定作用(图11)。X射线精细结构表征数据表明在NiFe合金中Ni和Fe之间存在电子相互作用,使得Fe原子周围电子密度升高,降低了水合肼中N—H键断裂的能垒。结合CO2-TPD对碱性位的数量以及强度进行定量,发现催化剂高的活性以及选择性主要归因于NiFe合金优异的N—H碱断裂能力和MgO载体的碱性位点的协同催化作用。近期,该课题组以CuFeMg-LDH为前体制备了Fe5C2-Cu/MMO催化剂,Fe5C2簇(约2 nm)被固定在Cu纳米颗粒(约25 nm)上形成了丰富的Fe5C2-Cu界面。在1 MPa的反应压力下,该界面催化剂表现出的CO转化率为53.2%,对长链醇选择性为14.8%(mol),时空产率为0.101 g·g-1·h-1,与其他Cu-Fe二元催化剂在3~8 MPa压力下的最佳时空产率水平相当。Fe5C2团簇与Cu纳米颗粒之间的协同相互作用是低压下高效催化生成长链醇的主要原因[5]。上述研究实例表明,对于能够进入Mg基LDHs层板的Fe、Co、Ni、Cu等金属元素,经LDHs前体路径是制备高分散、高稳定的负载型单金属、双金属催化剂的有效途径。

图11 NiFe-合金/MgO催化剂的高分辨透射电镜图像[(a)~(b)],高角环形暗场扫描透射电镜图像(c)及相应的Fe,Ni,Mg和O元素分布图(d)[61]Fig.11 Structural characterizationsof the NiFe-alloy/MgOsample:HRTEM[(a),(b)];High-magnification STEMimage with the EDS mapping(the scale bar is 20 nm)[(c),(d)][61]
3.2 负载型Pd基催化剂
Pd、Pt、Au等贵金属元素以及部分稀土元素不能进入或只能以很小的比例进入LDHs层板,可以采用传统浸渍、沉淀等方法将其负载在LDHs或MMOs上;或将含有活性金属元素的阴离子配合物插入LDHs层间,利用层板电荷分散性及可调性,实现金属元素阴离子配合物在层间的分散,进一步经焙烧还原过程获得高分散、高稳定的负载型贵金属催化剂。
He等[62]采用浸渍法制备了以MgO、Al2O3和MgAl-MMO为载体的负载型Pd基催化剂,在碳二选择性加氢反应中以MgAl-MMO为载体的Pd基催化剂表现出了最高的催化活性和选择性。结构表征结果说明,以MgAl-MMO为载体时Pd金属分散度最高(34.6%),且Pd具有较多的台阶、边角和缺陷结构。同时,MgAl-MMO载体表面碱性位能有效降低Pd的电子云密度,其具有最低的活化能,有利于加氢反应的进行;而采用单纯MgO载体时,表面强碱性位反而会提高Pd的电子云密度。构筑Pd基双金属催化剂可以进一步提高产物选择性,该课题组以含[PdCl4]2-的MgGaAl-LDH为前体,经气相共还原后获得了均匀分散的负载型双金属Pd-Ga/MgO-Al2O3催化剂,应用于乙炔选择性加氢反应(图12)。在相同的乙炔转化率(90%)下,相对于单金属Pd/MgOAl2O3催化剂55%的乙烯选择性,双金属Pd-Ga/MgO-Al2O3催化剂对乙烯的选择性高达82%。结构表征数据表明双金属Pd-Ga/MgO-Al2O3催化剂中活性组分颗粒尺寸更小,同时Ga的加入所引起的几何效应与电子效应促进了乙烯在催化剂表面的脱附,进而提高乙烯的选择性[9]。Yamaguchi等[63]通过在MgAl-LDHs上负载Pd纳米颗粒将其应用于环己酮肟选择性生成苯胺类化合物的反应中。在无助剂、还原剂和配体化合物加入的条件下,只是利用自身解离出来的H2,在热过滤过程中将Pd2+还原为Pd0。该催化剂对环己酮肟的催化加氢活性仍然能够保持苯胺类化合物80%左右的高收率,且在重复使用五次后,催化剂仍然能够保持较好的形貌和均一的粒径、无团聚现象。通过一系列的表征实验证实了Pd与MgAl-LDHs水滑石载体的协同催化使得环己酮肟通过一步脱水、脱氢生成苯胺。

图12 Pd插层MgGaAl-LDHs前体法合成双金属Pd-Ga/MgO-Al2O3催化剂示意图[9]Fig.12 Synthesis of the supported bimetallic Pd-Ga catalysts derived from Pd/MgGaAl-LDH@Al2O3 precursor[9]
3.3 负载型Pt基催化剂
负载型Pt基催化剂是过渡金属中最有效的烷烃脱氢催化剂之一,但是在缺乏改性剂的情况下,其对烯烃的选择性并不高,且烯烃在Pt表面再吸附产生焦炭而使催化剂失活。在多相催化体系中,金属-载体相互作用可以通过改变界面电荷转移、金属结构、分子吸附等,进而影响催化反应性能。水滑石的晶格限域效应,使镁基复合金属氧化物作为载体时充分发挥其金属-载体间的相互作用。
He等[64-65]制备了Pt/SnOx/Mg(Al)O@Al2O3催化剂应用于丙烷脱氢制丙烯,550℃下获得了高达30%的丙烷转化率及大于99%的丙烯选择性,反应运行10 d无明显失活。研究表明,Sn与Pt的强相互作用促进了Pt的高分散,并降低了Pt中心缺电子性,从而抑制了氢解反应且促进了丙烯的脱附。受限于水滑石主体层板Mg(Al)O晶格内的Pt在高温反应后依旧保持了原子级高分散,无聚集现象(图13)。类似地,Belskaya等[66]利用焙烧后的MgAl(Sn)Ox载体对氯铂酸进行吸附,MgAl(Sn)Ox在恢复为LDHs结构的同时将[PtCl6]2-嵌入LDHs层间,其在丙烷和正癸烷脱氢氧化反应中具有优异的催化性能。TEM、XPS和EXAFS等结构表征结果表明MgAl(Sn)Ox载体可以提高Pt的分散度,同时电荷从Sn转移到Pt上,进而提高了烯烃的选择性。

图13 高分散Pt/MgAl(Sn)O x催化剂的高分辨透射电镜图像及烷烃脱氢反应示意图[65]Fig.13 Schematic illustration and HRTEM images of Pt/MgAl(Sn)O x catalysts for dehydrogenation of alkanes[65]
除了利用Mg基LDHs晶格限域效应提高金属纳米颗粒的分散度和稳定性以外,还可以利用其固体碱催化剂的特点构筑双功能催化剂。Zhang等[67]结合浸渍法与焙烧还原-原位水合复原的方法制备得到MgAl-LDHs负载的Pt催化剂,应用于催化甘油氧化制甘油酸(图14)。研究发现通过调变Mg/Al比例,可以改变金属Pt纳米颗粒与载体间的相互作用,使得Pt纳米颗粒的价态逐渐升高,在界面处产生了大量缺电子的Ptδ+物种。正电性的Ptδ+促进了电负性Hδ-的脱除,提高了决速步α-Cδ+—Hδ-断裂的反应速率。同时,载体碱性的强弱对催化性能有显著的影响,当碱性较弱时,催化反应倾向于在中间位羟基上发生;而碱性过强时,甘油酸不容易脱附而发生过度氧化;适当强度的碱性有利于端位羟基的氧化并抑制产物的过度氧化,从而会获得较高的甘油酸选择性。近期,该课题组还以层状水滑石CuMgAl-LDHs为前体,制备了MgAl-MMO负载的PtCu单原子合金催化剂。其中Pt是以单原子状态分布于Cu纳米颗粒表面,实现了贵金属Pt的最高利用率及构筑Pt-Cu界面位点的最大化,显著提升了甘油氢解制备1,2-丙二醇的催化性能,其TOF值高达2.6×103 h-1,是目前报道的最高值[68]。

图14 MgAl-LDHs负载Pt催化剂催化甘油氧化制甘油酸反应机理示意图[67]Fig.14 A possible reaction pathway for the oxidation of glycerol over an Pt/MgAl-LDH catalyst[67]
3.4 负载型Au基催化剂
近年来,LDHs载体被视为多相醇氧化纳米Au催化剂的极佳载体[69]。2009年,Mitsudome等[70]报道了MgAl-LDHs负载纳米Au催化剂在无附加助剂条件下,以常压O2为氧化剂,催化液相芳基醇氧化生成醛、酮。研究表明较小的纳米Au颗粒(2~3 nm)和载体表面的碱性位是反应得以实现的关键因素。Zhang等[71]将Au负载在MgAl-LDHs上用于高效催化苯乙烯的环氧化反应,TOF值高达970.2 h-1。研究发现MgAl-LDHs的不同晶面结构会影响Au纳米粒子的沉积位置和颗粒尺寸,Au NPs优先负载在MgAl-LDHs的{1010}晶面,且与该晶面有较强的作用力,抑制了Au NPs在焙烧过程中的聚集现象,使其纳米粒子尺寸为2~3 nm(图15)。Wang等[72]通过离子交换和NaBH4还原两步法在MgAl-LDH上制备了粒径为1~5 nm的小尺寸Au纳米粒子。在对醇类氧化反应性能评价中发现,Au/LDH在非常温和的条件下,对多种伯醇和仲醇的氧化表现出优异的活性以及高的选择性。结构表征数据表明Au纳米粒子与LDH之间有强相互作用使Au带部分负电荷,带负电的Au纳米粒子有利于醇有氧氧化中分子氧的活化。此外,载体对于2-丙醇具有优异的脱氢能力。Du等[73]选取MgAl-LDHs为载体,制备了Au纳米颗粒仅为1.7 nm,而负载量却高达62.8%(质量)的Au/MgAl-LDHs催化剂。首先选用对苯二甲酸对N离子插层的MgAl-LDHs进行阴离子交换,扩大层间距离从而容纳Au前体。利用LDHs层板的限域作用抑制Au纳米颗粒的长大,达到小尺寸且均匀分散的目的,该方法为在载体上分散大量Au纳米颗粒而不发生严重聚集提供了一种新的方法。

图15 不同负载量的Au/MgAl-LDHs催化剂的高分辨透射电镜图像和结构示意图[71]Fig.15 Schematic illustration and HRTEM images of Au/MgAl-LDH with different loading[71]
An等[74]制备得到了具有不同表面碱密度及表面 羟 基 空 位 数 量 的Mg2Al、Zn2Fe、Co2Al、Zn2Al和Zn2Ga-LDHs作为载体负载Au纳米颗粒,实现了甘油仲羟基的高效转化制备二羟基丙酮。其中,Zn2Ga-LDHs负载纳米Au催化剂获得了高达72.9%±0.2%的甘油转化率以及63.8%±0.2%的二羟基丙酮选择性。对Au纳米颗粒的电子结构进行表征,发现除了表面Au0位点,还存在大量的界面MⅡ-O-Aun+(Au+与Au3+)活性位点。进一步关联LDHs表面碱性与反应TOF值以及界面MⅡ-O-Aun+与二羟基丙酮选择性,并对LDHs载体与LDHs负载Au催化剂进行原位红外研究,提出了LDHs表面碱性活化仲—OH与界面MⅡ-O-Au+位点活化仲C—H的协同催化机理(图16)。更为重要的是,对于Zn2Al-及Zn2Ga-LDHs负载Au催化剂,LDHs表面的羟基空位与界面的ZnⅡ-O-Au3+位点可进一步协同促进甘油的活化。结合同位素标记的研究方法,证实了LDHs负载纳米Au催化剂催化甘油选择性氧化制备二羟基丙酮的双重协同催化机理。Wang等[75]选用MgAl-LDHs为载体,采用浸渍-还原法制备了不同Au-Pd比例的负载型Au-Pd/MgAl-LDHs合金催化剂,其在可见光条件下光催化氧化苯甲醇制备苯甲醛。Au9-Pd1/MgAl-LDHs具有91.1%的转化率和99%的选择性,明显高于Au9-Pd1/TiO2催化剂的性能。Au-Pd合金之间的电子协同作用会导致电荷从Pd原子转移至Au原子,使Pd原子带正电荷,加强了其与O2之间的相互作用,有利于超氧自由基O2·-的形成。另一方面,MgAl-LDHs表面的碱位点活化苯甲醇分子,进而通过相互作用形成金属醇盐中间物,其更容易被O2·-氧化成为目标产物苯甲醛。Au-Pd合金与碱位点的协同催化作用促进了光催化选择氧化苯甲醇反应。

图16 水滑石负载纳米Au催化剂催化甘油选择性氧化制备二羟基丙酮的双重协同催化机理[74]Fig.16 Schematic illustration for the multiple synergies of supports in the selective oxidation of glycerol to dihydroxyacetone of LDHs supported Au catalysts[74]
由此可见,相对于采用传统的Al2O3、MgO为载体的负载型贵金属催化剂,以Mg基LDHs或MMOs为载体的优势在于:不仅提高了金属活性位的分散度,而且增强了金属与载体间的相互作用,从而实现了金属纳米催化剂高分散与高稳定性的统一;金属与载体间的强相互作用会导致界面电荷转移、金属结构改变、分子吸附调变等现象,进而影响催化剂的活性、选择性及稳定性;LDHs或MMOs具有丰富的Lewis酸碱位点,其与金属之间的协同效应可以提高反应的活性以及选择性。这些优势都使Mg基LDHs或MMOs在负载型催化领域有更多的发展潜力。此外,LDHs直接作为催化剂载体时,比表面积大,层板上富含羟基,其组成、形貌及结构更易于调变,也易于与其他材料杂化,尤其是与单层的LDHs纳米片复合可以展现出丰富的界面结构来提高其催化性能,是未来非常重要的研究方向。
4 镁基水滑石复合材料
稳定且环境友好的新材料是人类实现社会绿色可持续发展的关键之一。将Mg基LDHs与多种材料进行杂化或复合,如硫化物、氧化物、MOFs、石墨烯及其衍生物、碳纳米管等构筑镁基水滑石复合材料具有重要意义。该类复合材料在性能上互相取长补短,产生协同效应,使其综合性能优于原组成材料而满足各种不同的要求。
4.1 与纳米碳材料复合
纳米碳材料指分散相尺度至少有一维小于100 nm的碳材料,是过去30年间最为活跃的研究领域之一。富勒烯、碳纳米管、石墨烯等一系列碳质新材料的发现,带来了丰富的新结构和新应用。纳米碳材料具有特殊的几何结构,以sp2杂化为主的成键方式以及由此带来的优异的电学、热学、光学等特性。此外,纳米碳材料具有极高的稳定性,且易于大规模制备。上述优点恰好可以弥补Mg基LDHs自身的缺点,拓展其在能源、环境、催化等领域的应用。
在前文中提到,Mg基LDHs及衍生的MMO因具有高比表面积和丰富的表面碱位,是吸附增强型水-气转化反应的中温吸附CO2的重要材料之一。但是,仍存在吸附量相对较低、力学性能较差(容易浆化)等问题。将LDHs纳米片与石墨烯等碳材料复合后,可以改善LDHs的分散效果,避免纳米片的团聚,和单一LDHs组分相比,这种复合材料能在一定程度上明显增强其催化、吸附性能。Wang等[76]制备了单层LDHs纳米片与石墨烯复合材料(Mg3Al-LDH/GO)。由于氧化石墨烯(GO)富含官能团(羰基、羧基、羟基等)从而使其层板上带有负电荷,所以一些带有正电荷的阳离子就很容易进入到石墨烯层间,因此氧化石墨烯具有很好的离子交换性。利用该结构特点,将带正电荷的高分散的Mg3Al-LDH纳米单层溶液和带负电荷的氧化石墨烯纳米单层溶液混合,通过两种纳米粒子之间的静电自组装的方式,得到Mg3Al-LDH/GO复合材料。该复合材料在400℃条件下焙烧时吸附CO2效果最佳,在较宽的温度范围内都有很好的吸附CO2效果,吸附温度为60℃时,其吸附CO2效果最好,可达到1 mmol·g-1,而在中温区间内200℃时吸附效果最好。该复合方式使LDH纳米单层更好地分散在氧化石墨烯上,有效避免LDH纳米单层的重新组合与生长,而且充分利用了LDH的比表面积、活性中心、晶面取向性等。
Álvarez等[77]在碳纳米管(CNFs)上负载MgAl-LDHs,经焙烧复原后得到的固体碱催化剂在甘油与碳酸二乙酯交换反应中的催化活性与非载体催化剂相比提高近300倍。优异的催化性能归因于小尺寸的MgAl-LDHs暴露出了更多的碱性位点OH-,同时CNFs中的大孔也有利于反应物与碱性位点接触。Zou等[78]采用一步水热法制备了棉花状g-C3N4@NiMgAl-LDH类水滑石复合材料应用于水体中U(Ⅵ)的吸附,g-C3N4@Ni-Mg-Al-LDH对U(Ⅵ)的吸附量高达99.7 mg·g-1,明显优于Ni-Mg-Al-LDH(59.8 mg·g-1)和g-C3N4(31.1 mg·g-1)。g-C3N4具有丰富的非金属官能团(O—C O,C—O—C,—NH2等),LDH中具有丰富的金属-氧官能团(Mg-O,Al-O和Ni-O),这些官能团与UO2+2形成了较强的化学键或者其他铀复合物,使该类催化材料具有良好的吸附能力和化学稳定性(图17)。Guo等[79]用氮掺杂的碳纳米点来修饰MgAl-LDHs,并将其作为载体负载Au纳米颗粒,其在苯甲醇无碱、无溶剂选择性氧化成苯甲醛的反应中表现出了更好的催化性能。氮掺杂碳纳米点的引入提高了MgAl-LDHs的表面碱性,和Au纳米颗粒与载体之间的强相互作用,进而促进了苯甲醇的吸附和活化。
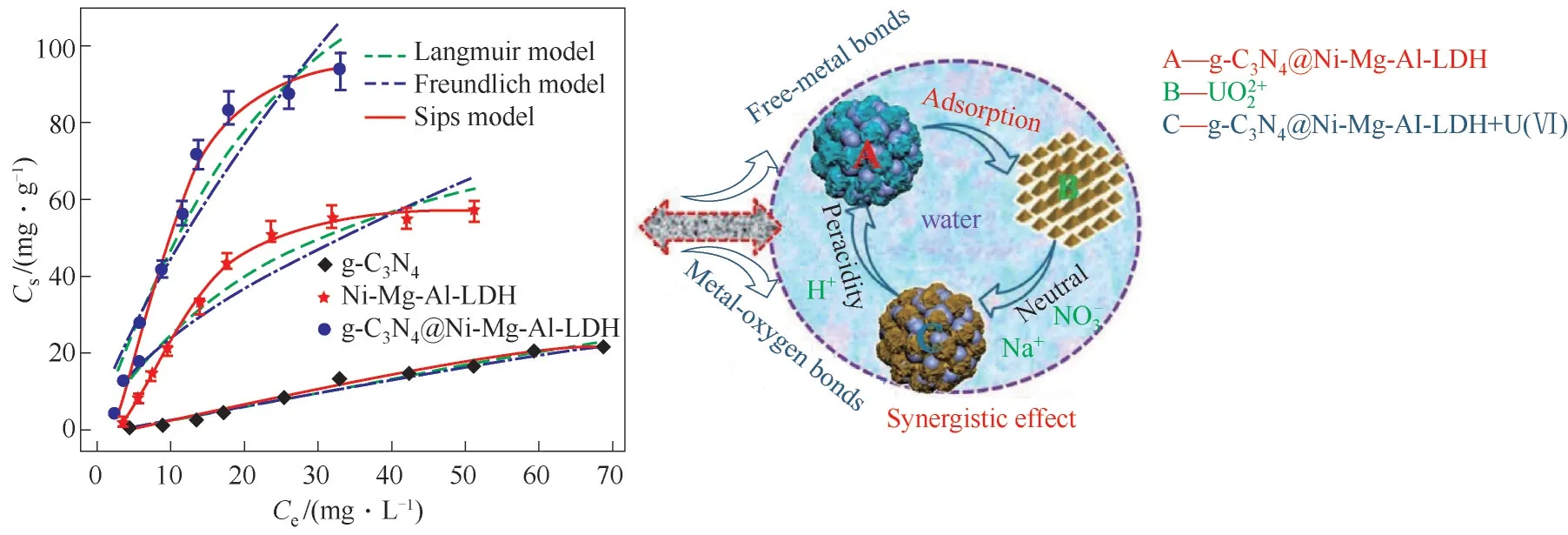
图17 g-C3N4@NiMgAl-LDH复合材料高效吸附水体中UO示意图[78]Fig.17 Schematic illustration for synergistic immobilization of U by g-C3N4@NiMgAl-LDH from wastewater[78]
4.2 与MOFs材料复合
金属有机框架材料(MOF)作为一类新型的晶态多孔材料,由于具有结构丰富,孔尺寸均一、可调,比表面积高,以及易实现功能化等特点引起科研工作者的高度关注,在催化、吸附和储存方面得到了广泛应用[80]。前文提到了LDHs基于其高比表面积、独特的层状纳米结构、丰富的官能团和离子交换能力,已经在吸附领域大放异彩。若将MOF与LDHs进行复合在理论上可以极大地提升材料的吸附性能。Chen等[81]成功制备了层状氢氧化物/金属有机骨架(LDH@MOF-76)复合材料,并用于水溶液中铀[U(Ⅵ)]的捕获。首先采用尿素法合成了CaMgAl-LDHs,再用水热法合成了CaMgAl-LDH@MOF-76。该复合材料能够快速有效地富集水溶液中的U(Ⅵ),表现出高的吸附容量(433.91 mg·g-1),远远高于单独的LDH(107.21 mg·g-1)和MOF-76(269.14 mg·g-1),且具有良好的循环使用性能。扫描电子显微镜(SEM)和比表面积分析结果表明所制备的复合材料显示出典型的多孔棒状结构,LDH@MOF-76材料表面特殊的一维孔道对U(Ⅵ)的吸附有很大的促进作用。基于Zeta电位和X射线光电子能谱学分析,U(Ⅵ)可能的去除机制包括两个关键途径:表面络合和静电吸引。铀酰离子和LDH@MOF-76上的羧酸(羰基和羟基)之间存在络合作用。此外,由镧系离子和均苯三甲酸组成的MOF-76具有沿c轴方向约6Å×6Å(1 Å=0.1 nm)的一维通道,其孔径完全能够捕获线性铀酰阳离子。该工作为废水中铀(Ⅵ)的去除和Mg基LDHs的拓展应用提供了新的研究思路,并且具有好的应用前景。
以MOF为前体制备多孔碳材料是MOF材料的另一个重要应用领域,但是其经热解碳化制备碳材料时,MOFs颗粒容易聚集,且原有孔道结构塌陷,极大地限制了活性位点的暴露。LDHs表面的金属阳离子处于一种不饱和的配位状态,利用其与MOFs有机配体之间的金属-有机配位作用,可以实现MOFs晶体阵列的导向生长,得到均匀有序的LDHs@MOFs核壳式复合材料,进而经过高温碳化处理得到结构新颖的二维多孔碳骨架材料。Li等[82-83]将ZIF-67纳米晶阵列定位生长在CoAl-LDHs表面,进而通过高温碳化处理得到一种结构新颖的蜂窝状碳骨架材料。所制得的二维碳骨架材料拥有优异的氧还原反应(ORR)性能,其起始电位和半波电位高达0.94 V(vs.RHE)和0.85 V(vs.RHE),同时具有优异的甲醇耐受性和循环稳定性。从HRTEM和SAED谱图中可以看到碳化过程导致LDHs@MOFs中的有机物转变成多孔石墨化碳,而单纯ZIF-67晶体以球状纳米颗粒形式存在,并且碳化后无法形成多孔碳纳米结构,而是堆积在一起的碳颗粒。CoAl-LDHs主体层板中Co2+的含量及分布对所制备的碳材料的结构及形貌具有显著影响。当Co含量较低时,无法提供足够的配位反应位点来锚定ZIF-67纳米晶体;而过多的Co导致过量的ZIF-67包覆在LDHs表面,无法形成有序多孔碳骨架。进一步,该课题组以LDHs@MOFs为依托,在MOFs中引入Co、Zn两种过渡金属,通过调节Co/Zn的摩尔比,实现了对所制备碳骨架材料的形貌、杂原子(N,Co)掺杂量、石墨化程度以及比表面积和孔结构的调控。在LDHs@ZIF-67-CoZn前体中,Co离子有助于催化形成高石墨化的碳,提高所制备碳材料的导电性;Zn离子有助于提高碳材料中N的掺杂量,为ORR反应提供足够的活性反应位;而LDHs作为模板定位生长基元,为二维多孔有序碳材料的制备提供支架。
基于LDHs的结构特点,将MOF化合物与LDHs复合制备结构有序的复合材料,可以有效解决MOF材料在水蒸气或酸性气体条件下易发生结构破坏、高温热解过程中金属会发生团聚和骨架的塌陷、孔道结构狭窄、机械强度较差等问题;LDHs层板上过渡金属离子的可调变性和MOFs种类的多样性,可以设计制备出种类丰富LDHs@MOFs复合纳米材料,增强对其结构、组成、相貌的可调控性,进而获得性能强化的二维纳米复合材料。
4.3 与金属硫化物、氧化物材料复合
二维纳米片层状MoS2是过渡金属硫化物材料家族的典型代表,每层MoS2由Mo原子夹在两层S原子间形成三明治的结构,层与层间由范德华力连接,这种结合力较弱可以被剥离成单层或几层的纳米片层结构。基于该特殊结构,MoS2及其复合材料在催化、能量储存、传感器等方面有广阔的前景[84-86]。将同样具有层状结构带正电荷的LDHs与带负电荷的MoS2复合,可以构筑高效的纳米复合光催化材料。具有合适价导带电位的两种层状结构复合形成的异质结结构可克服单一光催化剂固定带隙的局限,可调节光生电荷转移过程,在光催化CO2还原等反应中表现出优异的催化性能。Qiu等[84]将带正电的超薄CoAl-LDH和带负电的超薄MoS2材料通过静电作用组装形成具有异质结结构的LDH/MoS2纳米复合材料光催化剂,通过异质结诱导了电子竞争过程,成功调节了CO2还原产物中CO/H2的比例。在可见光照射(λ>400 nm)条件下,仅通过在0.2~1.5 mg·ml-1范围内调控催化剂浓度,将CO2还原生产合成气(H2∶CO),实现产物比例在1.3∶1~15∶1范围的精准调控。HRTEM和Zeta电位测试证明了LDH/MoS2异质结结构成功形成。通过Mo的K边XANES曲线、EXAFS曲线以及XPS结果表明该材料的能带结构和局部配位结构均有变化:电子从LDH向MoS2转移使Mo价态升高,且LDH/MoS2中形成了Mo-O壳层,表明O原子通过化学吸附到LDH/MoS2晶格中形成LDH/MoOSx的结构,进而增强了两组分间的相互作用。稳态光致发光光谱表明提高LDH/MoS2催化剂的异质结浓度可以有效转移光生电荷,当异质结浓度增加时,光生电荷分离效率提升。
利用LDHs独特的层状结构和高的离子电导率等特性,将其与MoS2构筑复合材料,在能量存储方面也引起了人们的广泛关注。Xu等[85]同时将碳纤维、MoS2、MgAl-LDH与石墨烯进行自组装制备了轻便(<0.25 mg·cm-2)、超 薄(<0.8 mm)、高 效 的CFCMoS2/MgAl-LDH-GO复合材料(图18)。将其应用于超级电容器,展现出了非常高的能量密度,功率密度为1500 W·kg-1时的能量密度为326.54 W·h·kg-1。甚至功率密度升至112 kW·kg-1,依旧能保持279.56 W·h·kg-1的能量密度[85]。MgAl-LDH-GO本身的电荷转移电阻较低、离子扩散速率较高。但将MgAl-LDH-GO与CFC-MoS2复合后,稳定的结构、增强的电化学动力学和快速的离子转移使CFC-MoS2/MgAl-LDH-GO非对称超级电容器获得了具有良好循环稳定性的高能量和功率密度。但是,由于MgAl-LDH的电化学性能较差,将其与MoS2等过渡金属硫化物复合的研究十分有限。研究更多的是选用电化学性能较好的Co基、Ni基LDHs(例如CoAl-LDHs、NiFe-LDHs),尤其是通过对超薄LDHs/MoS2复合材料的界面结构、缺陷结构、电子结构、晶相结构等的微观调控,是提高MoS2本征催化活性以及整体催化性能更适宜的手段[86]。

图18 CFC-MoS2阳极、MgAl-LDH-GO阴极及非对称超级电容器组装示意图[85]Fig.18 Schematic illustration of CFC-MoS2 anode,MgAl-LDH-GOcathode and the assembled asymmetric supercapacitor[85]
除了与上述过渡金属硫化物复合,Mg基LDHs与其他一些金属氧化物(如CuO、Co3O4、CeO2、TiO2、MnO2等)复合的材料也逐渐活跃起来。Cu2+、Ni2+、Co2+、Ti4+等金属离子满足LDHs的八面体构筑原则,因此可以直接引入到LDHs层板结构构筑二元、三元、多元Mg基LDHs,进一步焙烧后获得CuO/MgO、CeO2/MgO、MgO/TiO2等复合材料。利用MgO的碱性及CuO等金属氧化物的催化性能,其作为双功能催化材料广泛应用于CO氧化反应、CO2转化、光催化反应等领域[87-89]。而MnO2、CeO2等则一般需要自组装、共沉淀、浸渍法等方法制备复合材料。Yamini等[90]采用静电自组装法将带正电荷的MgAl-LDH纳米片和带负电荷的层状δ-MnO2纳米片组装制备了有序的纳米杂化材料。在此基础上,将组氨酸(His)两性离子作为连接剂置于带正电荷的MgAl-LDH纳米片和带负电荷的层状δ-MnO2纳米片的层间空隙中,构筑了层状有机-无机杂化纳米结构,研究了其对水溶液中重金属阳离子Pb(Ⅱ)和As(Ⅴ)和含氧阴离子的共脱除性能[90]。该材料在30 min内即可达到吸附平衡,对As(Ⅴ)和Pb(Ⅱ)的吸附量分别为632.91和584.80 mg·g-1。在吸附过程中,MnO2的高比表面积和负电荷促进了带正电荷的重金属离子通过静电吸引快速迁移到LDH/His/MnO2表面。组氨酸的羧基和胺官能团也有利于重金属的高效吸附,与羧基配位的重金属离子可以进一步吸附到LDH/His/MnO2样品上。而MgAl-LDHs表面的正电荷通过静电吸引作用促进了As(Ⅴ)含氧阴离子向LDH/His/MnO2外围的快速迁移,此外组氨酸的氨基官能团也有利于含氧氧阴离子的高效吸附。因此,MgAl-LDH/His/MnO2复合材料利用其物理吸附和化学吸附的协同作用,高效去除了水中的氧阴离子[As(Ⅴ)]和重金属[Pb(Ⅱ)],该研究提供了一条提高吸附效率的有效策略。
4.4 与磁性材料复合
粉体水滑石催化剂在液相催化中的缺点是难以从固/液混合物中分离出来,采用传统的离心或过滤等物理方法,耗时耗能还会造成催化活性组分的流失,因此合成高性能且可通过外加磁场回收循环利用的磁性催化剂体系具有重要的环境和经济意义。在常见的磁性纳米材料中(Fe、Co、Ni的金属氧化物等),最常用的是Fe3O4,其制备方法简单、稳定性好、粒度可控、生物毒害作用小,在环境、材料、催化等领域应用广泛。
Mi等[91]通过一步共沉淀法制备了具有超顺磁特性、尺寸均一的多级核壳结构复合物Fe3O4@MgAl-LDH,并将其作为载体负载纳米Au粒子用于1-苯乙醇氧化反应(图19)。载体壳层MgAl-LDH的碱性位与活性组分纳米Au的协同作用使该催化剂在较低反应温度、以氧气为氧化剂且无须添加碱助剂的反应条件下具有高催化活性和选择性。进一步,将镁基LDHs与磁性材料如Fe3O4复合后再用于吸附水体污染物,不仅能进一步提高材料的吸附性能,同时还能利用磁性实现催化剂的快速分离、回收再利用。Yan等[92-95]制备了具有磁性的Fe3O4/LDH/氧化石墨烯复合材料,用于吸附水溶液中的重金属离子等污染物。水体中的重金属如Pb2+、Cu2+和Cd2+与复合材料表面官能团络合,进而生成金属氢氧化物和金属碳氧化物的沉淀,利用水滑石的“记忆效应”,重金属替换LDHs层板Mg2+完成吸附过程。郝瑞霞等[96]采用共沉淀法制备了磁性Mg/Al-LDHs以提高粉末状LDHs吸附剂的工程应用性。结果表明,在Fe3O4与Mg2+物质的量比为0.02、Mg/Al物质的量比为2时磁性Mg/Al-LDHs的吸附除磷效果最好,平衡吸附量可达49.60 mg/g,比饱和磁化强度为2.78 emu/g,能够实现吸附剂的快速高效磁分离回收。

图19 磁性Fe3O4@MgAl-LDH@Au催化剂的合成过程示意图[91]Fig.19 The synthetic strategy for an Fe3O4@MgAl-LDH@Au catalyst[91]
Mg基LDHs复合材料兼具LDHs和其他功能材料的双重优点,尤其是在污染物吸附方面,不仅可以弥补Mg基LDHs材料应用于吸附分离领域存在的缺陷,还可以利用两组分间的协同作用,进一步强化复合材料的吸附分离性能。通过更为简单、温和的方法制备出功能更强大、绿色环保、用途更为广泛的新型LDHs纳米复合材料是推向实际应用的重要途经。
5 结 论
近年来,镁基水滑石催化材料的研究已经取得了一定的进展,正受到越来越广泛的关注。本文重点综述近几年来基于镁基水滑石催化材料的研究进展,并总结其结构调控规律,为高性能镁基水滑石催化材料的设计和合成提供了一定的依据。尽管镁基水滑石在诸多催化反应中都表现出良好的应用前景,但仍存在一些问题需要解决,诸如催化剂的活性位点的不明确,特别是焙烧后的复合金属氧化物和镁基水滑石复合材料等较为复杂的催化剂,影响了对其催化机理的研究。因此,深入理解镁基水滑石结构特点的科学本质,揭示催化机理以及催化剂之间的协同催化作用,有针对性地精准设计并可控制备高活性、高选择性以及高稳定性的镁基水滑石催化材料,加快其在催化领域的工业化应用,是盐湖镁资源综合开发循环利用的研究重点。
