溶剂极性响应型丁基-环四联吡啶提锂分子的合成及性能研究
2021-06-30李恩泽叶培远王亚欣康锦阴彩霞程芳琴
李恩泽,叶培远,王亚欣,康锦,阴彩霞,程芳琴
(1山西大学资源与环境工程研究所,山西太原030006;2山西大学分子科学研究所,山西太原030006)
引 言
锂(Li)因其独特的物理化学性质已被广泛应用于核能、航空航天、新能源、智能制造等行业,锂作为新型能源和战略性资源的重要地位也日益凸显[1-2]。我国锂资源储量丰富,其中盐湖卤水锂资源占80%以上。近年来,盐湖卤水锂资源的提取和开发利用已逐渐受到重视和关注[3-4]。但是,我国盐湖卤水具有高镁锂比的特点,镁和锂在元素周期表中处于对角线位置,二者溶液性质和极化能力相似,锂离子的选择性提取难度大[5-9]。
目前国内外已经发展了系列的盐湖提锂的技术,比如沉淀法、溶剂萃取法、吸附法、膜法、电渗析法等[10-12]。其中溶剂萃取法收率高、设备简单、易于实现连续操作和自动化控制,且适用于高镁锂比盐湖卤水[13-15]。多年来,工业生产中常采用磷酸三丁酯(TBP)作为萃取剂提取盐湖卤水中的锂离子,但此过程中必须加入大量FeCl3作为协萃剂,随后采用浓度为6~9 mol/L的盐酸溶液进行反萃,得到锂产品[16-20]。这种多级串联的萃取-反萃工艺流程复杂,协萃剂的加入会提高生产成本且影响产品纯度,高浓度盐酸的使用增加了操作危险性。如果能开发一种新型提锂分子,在无须协萃剂下能够选择性地与锂离子直接作用,且只需改变外界条件就能使锂离子脱除,既能节约成本减少操作危险性,也符合现代化工“简单化、绿色化”生产的新要求。基于此想法,课题组前期设计合成了具有光响应的提锂分子,实现在紫外光下与锂离子选择性“络合”,而在可见光下“释放”锂离子,且在不同光照下有颜色变化,具有可视化的效果[21-22]。然而光响应提锂分子仅适用于透明设备,限制了其实际应用。
环状联吡啶类化合物分子由多个吡啶亚基通过碳桥或者直接相连组成,具有空腔结构和多个N原子供体,可通过“离子-偶极”作用和碱金属离子形成稳定络合物,同时通过改变环状的空腔尺寸大小,可实现对特定金属离子的选择性识别[23-26]。Ogawa等[27]以6,6-二溴-2,2-联吡啶为原料设计合成了双丁基四联吡啶分子,发现该分子对锂离子具有较高的选择性识别能力。此外,环内与吡啶亚基相连的两个H原子在不同极性溶剂中会发生质子转移,溶液颜色由在非极性溶剂(二氯甲烷)中的红色向极性溶剂(甲醇)中的无色发生转变,表现出溶剂极性响应性。
本文在前期报道的基础上,同样合成具有溶剂极性响应特性的单丁基四联吡啶分子。该分子在非极性溶剂中时,环内H原子与相邻N原子形成共轭结构,当该分子处于极性溶剂时,H原子转移至与氰基相连的桥碳原子上,即发生溶剂极性响应的“异构互变”,此时环内空腔空出,能够与锂离子发生络合作用。文中进一步分析了该丁基四联吡啶分子的光谱性质和对锂离子的识别特性,同时采用液体支撑膜的方法对锂离子的提取性能进行了初步研究。
1 实验部分
1.1 实验试剂和仪器
药品试剂:氢化钠(60%分散于矿物油),氰乙酰胺(98%),溴代正丁烷(>98.0%),1,4-二氧六环(≥99.5%),2-硝基苯辛醚(98%),固体高氯酸锂(99.99%),固体高氯酸镁(99.99%),无水氯化锂(99.99%),无水氯化镁(99.99%),均购自上海阿拉丁生化科技股份有限公司;石油醚(分析纯),N,N-二甲基甲酰胺(DMF,无水,>99.9%),6,6'-二溴-2,2'-联吡啶(97%),二氯甲烷(分析纯),1,2-二氯乙烷(99%),无水乙醇(分析纯),均购自上海麦克林生化科技有限公司。整个实验过程中所用水为Milli-Q水(电阻率18.2 MΩ·cm,实验室自制)。
仪器设备:UV-26001型紫外可见分光光度计(苏州岛津仪器有限公司);600 MHz核磁共振仪(德国KRUSS公司),NexION 350型电感耦合等离子体质谱仪(美国PerkinElmer公司),IKARV10旋转蒸发仪(德国IKA仪器公司)。
1.2 6-丁基-3,6-二氰基-环四联吡啶的合成
6-丁基-3,6-二氰基-环四联吡啶分子(C)的合成路线如图1所示,具体合成步骤如下[28]。

图1 6-丁基-3,6-二氰基-环四联吡啶分子的合成路线及不同溶剂极性下分子结构的可逆变化和络合锂离子示意图Fig.1 The synthetic route of butyl tetrapyridine,reversible changes of molecular structure under different solvent polarity and schematic complexion of lithium ion
(1)化合物B的合成:室温下分别将2.0 g氢化钠(表面矿物油采用石油醚清洗),1.9 g 6,6'-二溴-2,2'-联吡啶和2.0 g氰乙酰胺溶解或者均匀分散到20、20和10 ml无水DMF中。随后在氮气气氛下,依次将氰乙酰胺DMF溶液和6,6'-二溴-2,2'-联吡啶DMF悬浊液缓慢滴加入放有氢化钠溶液的100 ml三口烧瓶中,在120℃下搅拌回流反应6 h,随后加水终止反应。抽滤并用水和丙酮清洗得到一步产物B,60℃下真空干燥12 h后密封待用。
(2)化合物C的合成:在50 ml三口烧瓶中放入0.2 g石油醚清洗后的氢化钠,然后室温和氮气气氛环境下依次加入10 ml无水DMF和0.1 g一步产物B,60℃下搅拌反应2 h后,加入溴代正丁烷,温度升至110℃后持续搅拌回流反应3 h,之后加水终止反应,抽滤水洗并干燥后得到固体混合物。将干燥后的固体混合物溶解于二氯甲烷,以二氯甲烷作为淋洗剂,过硅胶柱,承接第一层样品,减压旋蒸后于60℃下真空干燥12 h,得到最终产物C,固体粉末状,产品纯度99%。C难溶于水,易溶于甲苯、1,2-二氯乙烷、2-硝基苯辛醚等非极性有机溶剂。
1.3 分子结构和性能表征
反应过程中的中间化合物和最终产物C的分子结构采用核磁共振氢谱(1H NMR)和傅里叶变换红外(FT-IR)进行表征,产物C与阳离子的络合结构和离子选择性识别性能采用电感耦合等离子体质谱和紫外-可见光谱进行分析。
1.4 金属离子的跨膜传输
文中所得产物C对锂离子的提取性能采用跨膜传输的方法进行研究,该方法可实现金属离子的连续传输,无须萃取和反萃的交替操作,具体装置和过程如图2所示。装置中包含原料相、接收相和膜相,膜相采用液体支撑膜[29-31],油水界面处锂离子的传输过程如下:第一步是在极性含锂水溶液和非极性含提锂分子溶液的界面处,提锂分子因接触到界面极性水相,质子氢从内核向外环移动,暴露出氮原子位点,络合锂离子;第二步是络合有锂离子的提锂分子会随着搅拌从界面向非极性油相的体相转移,由于体相溶剂为非极性,质子氢会返回到原来位置,屏蔽氮原子位点,释放锂离子;第三步,由于氯化锂在非极性油相中溶解度极低,锂离子会在浓度梯度的驱动下经由新的油/水界面转移到接收相,完成锂离子的提取。

图2 离子的跨膜传输装置及过程Fig.2 Schematic illustration of cation transmembrane transport devices and processing
在实验过程中,左侧水溶液为原料相(体积135 ml,Li+或Mg2+浓度为500 mg/L),右侧水溶液为接收相(体积110 ml,Li+或Mg2+浓度为20 mg/L),中间为膜相。液体支撑膜的制作是首先将一张直径为50 mm的聚丙烯微孔膜(孔径0.45μm)在浓度为1×10-2mol/L的丁基-环四联吡啶有机溶液中浸泡12 h,有机溶剂为1,2-二氯乙烷和2-硝基苯辛醚的混合液,二者体积比为4∶1。浸泡后的膜使用滤纸小心擦除表面多余液体,膜内约含有40μl液体膜溶液(称重法计算),随后将其安装在装置中进行离子跨膜传输。原料相和接收相内搅拌速度均为1000 r/min,搅拌一定时间后,两边均取水溶液1 ml,稀释特定倍数后用电感耦合等离子体(ICP-AES)进行相关离子含量的测定。
跨膜传输结束之后,分别通过式(1)和式(2)计算分配系数Kd和选择性分离因子α。

式中,m接和m原分别表示跨膜传输结束后某单一金属离子在接收相中增加的质量和原料相中剩余的质量;Kd(Li+)和Kd(Mg2+)分别表示Li+和Mg2+的分配系数。
2 实验结果与讨论
2.1 6-丁基-3,6-二氰基-环四联吡啶的分子结构
2.1.1 FT-IR表征 为了说明由化合物B到化合物C发生了丁基取代反应,分别对两种化合物进行了FT-IR测试,结果如图3所示。结果表明,化合物B在2185 cm-1处表现出特定的—CN伸缩振动峰,且该伸缩振动峰的强度远高于普通的—CN伸缩振动峰,这是由于它与化合物B共轭结构的相连而增强了峰强度。化合物B经过单丁基取代后生成的化合物C部分保留了化合物B的共轭结构,因此在2185 cm-1处表现出与化合物B相同的—CN(共轭结构)的伸缩振动峰,但丁基取代改变了部分的共轭结构,导致化合物C在2242 cm-1表现出一个较弱的—CN(非共轭结构)的伸缩振动峰,上述结果符合化合物C的结构特征。

图3 化合物C和化合物B的FT-IR谱图Fig.3 FT-IRspectrumof compound C and compound B
2.1.21H NMR和高分辨质谱表征 为了进一步确定产物分子结构,对化合物C分子进行了核磁共振氢谱和高分辨质谱分析,核磁共振氢谱测试溶剂为氘代DMSO,结果如图4所示。经过分析,各氢原子化学位移及积分氢原子数结果如下:1H NMR(600 MHz,DMSO-d6),δ=15.48(s,1H),δ=8.03(s,4H),δ=7.91(d,2H),δ=7.85(t,2H),δ=7.55(d,2H),δ=7.46(d,2H),δ=1.19(s,6H),δ=0.74(s,3H)。该结果与产物C分子中氢原子的位置及数目完全对应,证实了产物分子结构的正确性及高纯度。此外,高分辨质谱图中(图5)在m/z=443.20处存在明显的化合物C的分子离子峰,证明化合物C分子结构的准确性。

图4 化合物C的1H NMR谱图Fig.4 1H NMRspectrumof compound C
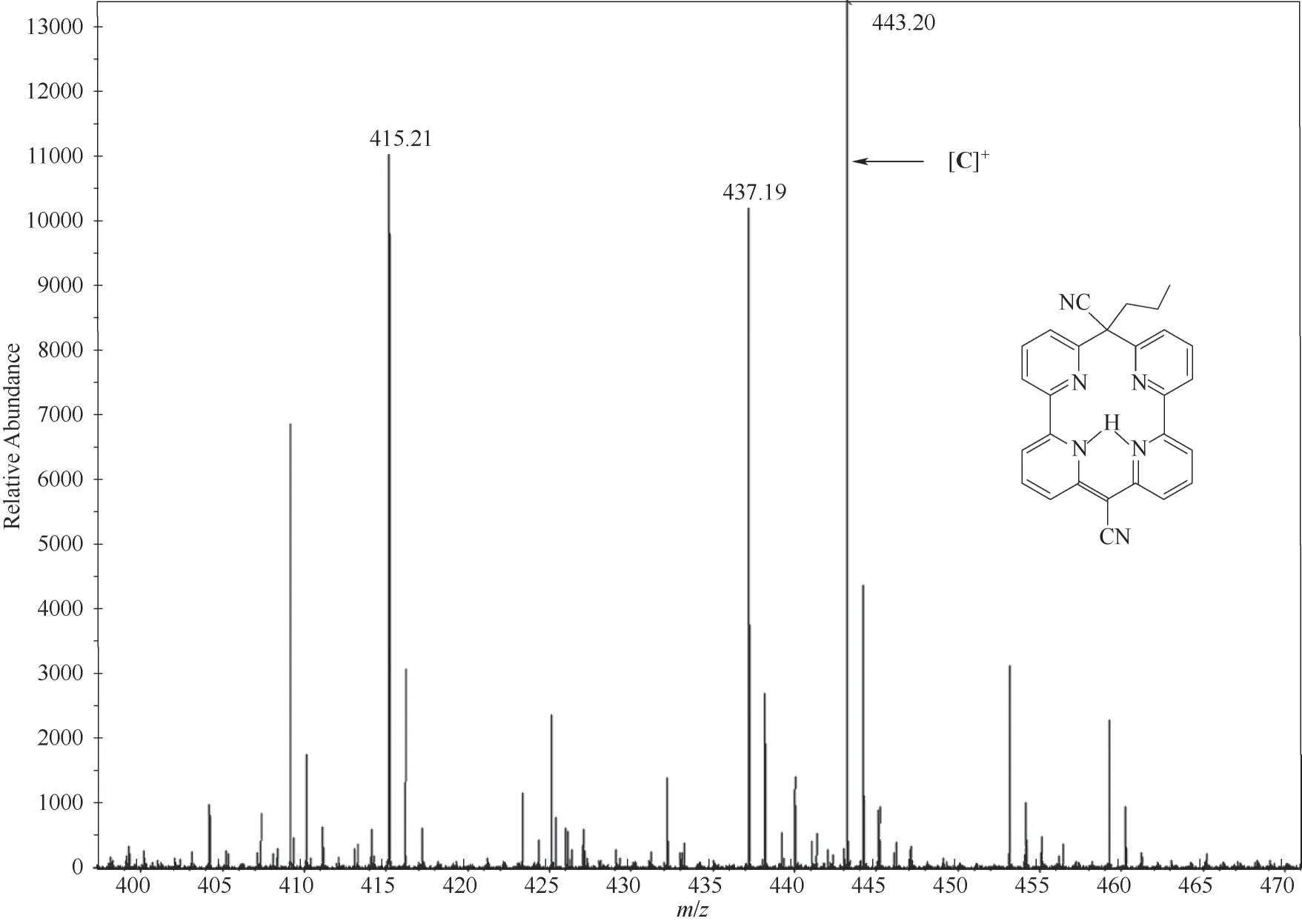
图5 化合物C的高分辨质谱图Fig.5 HRMSspectrumof compound C
2.2 6-丁基-3,6-二氰基-环四联吡啶的溶剂极性响应性
为了考察丁基-环四联吡啶分子对溶剂极性的响应性,首先将该分子溶于极性较弱的1,4-二氧六环(1×10-4mol/L),随后向其中逐渐加入微量的纯水,以逐渐增强溶剂的极性,同时采用紫外-可见光谱测试监测分子结构的变化。紫外-可见光谱的结果(图6)显示,随着微量纯水的加入,紫外-可见光谱上357 nm处的吸收峰有明显的降低。这是因为随着溶剂极性的增加,极性能够诱导环内质子转移至环外,导致丁基-环四联吡啶分子从化合物C的共轭结构向化合物D的非共轭结构逐渐发生转变(分子结构变化如图1中所示),在紫外-可见光谱上则体现为357 nm处吸收峰的降低。
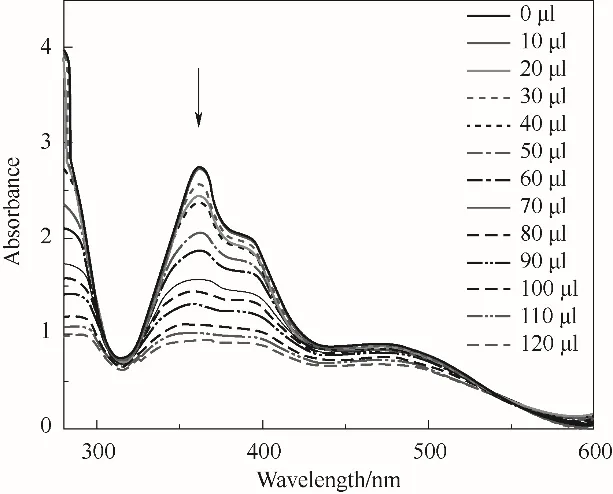
图6 化合物C的1,4-二氧六环溶液中逐渐加入微量纯水的紫外-可见光谱变化Fig.6 The UV-Vis absorption spectral change of compound C by addition of water in 1,4-dioxane
2.3 6-丁基-3,6-二氰基-环四联吡啶对锂离子的选择性
盐湖卤水中镁锂分离是锂离子提取的关键,对锂离子的选择性是提锂分子的重要特性。本实验同样通过紫外-可见光谱的测定考察化合物C对锂离子的选择性,结果如图7所示。结果表明,锂离子存在的情况下,在357 nm处的吸光度有明显降低,而镁离子存在的情况下,与无金属离子存在的光谱基本一致。说明锂离子与化合物C能够发生络合,而镁离子则不能。锂离子与化合物C的络合结构如图7中小图所示。这主要是由于化合物C特殊的环状结构,该环状结构柔韧性较差,而金属离子只能在环中心与周围的四个氮原子配位,因此能否与某种金属离子络合,环的空腔大小起决定作用[27]。化合物C的空腔较小,只有离子半径小的锂离子才能进入空腔内发生配位络合,而半径相对较大的镁离子则不能,表明化合物C对锂离子具有较强的选择性。
图8给出了极性溶剂中(1,4-二氧六环和水的等体积混合液)Li+或Mg2+存在时化合物C的高分辨质谱图。如图8(a)所示,在m/z=449.21处存在一个明显的离子峰,表明化合物C和Li+形成了稳定的络合物;然而图8(b)中只存在化合物C的分子离子峰(m/z=443.20)但不存在化合物C和Mg2+所形成的络合物的离子峰(m/z=446.16),表明化合物C只能选择性地与离子半径较小的Li+形成稳定的络合物,而不能和离子半径较大的碱土金属离子Mg2+发生络合反应。
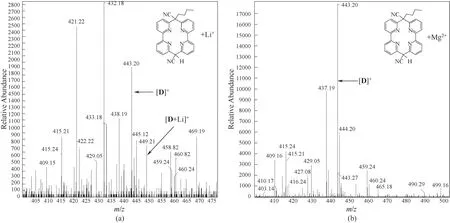
图8 Li+或Mg2+存在时化合物C在1,4-二氧六环-水溶液中的高分辨质谱图(化合物C的浓度为1×10-6 mol/L,1,4-二氧六环和水的体积比为1∶1)Fig.8 HRMSspectrum of compound C in the presence of Li+or Mg2+in 1,4-dioxan aqueous solution(the concentration of compound Cwas 1×10-6 mol/L,the volume ratio of 1,4-dioxane to H 2Owas 1∶1)
2.4 锂离子选择性跨膜传输性能
采用跨膜传输的形式,研究得到丁基-环四联吡啶分子对锂离子的选择性提取性能。图9给出了Li+和Mg2+在跨膜传输过程中接收相离子浓度增加量随时间的变化情况,表明Li+能够明显地从原料相向接收相迁移,而Mg2+基本不迁移,同样说明丁基-环四联吡啶分子对锂离子具有选择性提取性能。这个传质过程主要是由于采用的液体支撑膜相中溶剂为1,2-二氯乙烷和2-硝基苯辛醚的混合液,极性较弱,而原料相和接收相均为水溶液,极性较大。在原料相和膜相的界面处,部分丁基-环四联吡啶分子以化合物D的结构形式存在,该结构能够在界面处与原料相中的锂离子络合,随后络合物迁移至膜相本体,由于膜相极性较弱,化合物D的分子结构可逆转变成化合物C的结构形式,此时锂离子脱除,在浓度驱动下锂离子进入接收相,实现锂离子的跨膜传输。此外,通过测定跨膜传输完成后各离子在接收相中的质量增量和原料相中的剩余质量(表1),根据式(1)和式(2)计算可得,Li+和Mg2+的分配系数分别为0.056和0.001,则该实验条件下对Li+的选择性分离因子α为56,进一步说明所得丁基-环四联吡啶对锂离子具有较高的选择性提取性能。由于丁基-环四联吡啶在水中的溶解度极低,无进入水相所造成的损耗,实验中液体支撑膜可持续运行24 h以上,稳定性较好。目前锂离子的传输效率较低,在今后的工作中可通过增大两侧压差,或者通过加电形成电压差来促进锂离子的传输速率。

图9 Li+和Mg2+在跨膜传输过程中接收相阳离子浓度增加量随时间的变化Fig.9 The increment of the receiving phase cation concentration varies against time during the transmembrane transmission of Li+and Mg2+

表1 跨膜传输后Li+和Mg2+的分配系数和选择性分离因子Table 1 The partition coefficient and selective separation factor of Li+and Mg2+after membrane transport
3 结 论
本文成功合成了具有溶剂极性响应性的丁基-环四联吡啶提锂分子,在极性和非极性溶剂中该分子通过质子转移能够可逆异构互变。与镁离子相比,该提锂分子能够与锂离子进行选择性络合,这主要是由于环四联吡啶的内环尺寸大小与锂离子大小尺寸匹配,锂离子处于环中心与周围的四个氮原子配位。通过将丁基-环四联吡啶分子负载于极性较弱的液体支撑膜相内,锂离子能够选择性地进行跨膜传输,跨膜效果远高于镁离子。由于丁基-环四联吡啶分子的合成路线复杂,使用成本较高,进一步优化合成路线或采用其他廉价原料作替代,提高收率,降低成本,将成为下一步的工作重点。
