PMP22基因重复突变导致的腓骨肌萎缩症1例
2021-06-30林文鑫王春莉郑必霞周玮郑帼张霞黄艳军
林文鑫,王春莉,郑必霞,周玮,郑帼,张霞,黄艳军
腓骨肌萎缩症(CMT)是周围神经病变引起的遗传病,CMT1型是最常见的亚型,约占所有病例的70%,常在20岁之前发病[1]。症状轻微者可仅有感觉不适甚至无症状,而严重者可导致瘫痪或残疾。目前已有超过100个基因被确定与CMT及其相关疾病的病因相关[2]。基因检测已成为对该病早期诊断的重要工具。现报告1例由PMP22基因1至5号外显子单倍重复导致的CMT患儿的临床资料如下。
1 临床资料患儿,女,11岁9月。因“步态异常7年余”于2019年9月17日至我院门诊就诊。患儿智力发育正常,运动发育较一般同龄儿稍落后,2岁龄可独走,约4岁龄发现步态异常,表现为跨域步态,右下肢明显,进行性加重,无感觉异常,否认有足部外伤病史。至其他三级医院行脊柱正侧位片及EMG分别提示脊椎侧弯及周围神经损害,遂至我院神经科门诊就诊。父亲体健,母亲和外婆均有不同程度弓形足及步态异常。入院查体:脊柱侧弯。弓形足,双足背屈无力,跖屈弱。双下肢肌力Ⅲ~Ⅳ级,双上肢肌力正常,四肢肌张力正常,腱反射未引出,病理征阴性。实验室检查:脊柱正侧位片示腰椎轻度侧弯,S1椎体可见隐裂。全脊柱MRI平扫示脊柱侧弯,胸腰段为著。头颅MRI平扫+MRA未见明显异常。EMG示上下肢多发运动、感觉周围神经损害(脱髓鞘合并轴索损害),其中正中神经运动传导速度9.5 m/s,尺神经运动传导速度8.3 m/s。综合检查结果及家族史高度提示遗传性周围神经病,进一步行全外显子组测序(北京全谱医学检验实验室)显示,患儿17号染色体存在大片段CNV,是位于chr17:14095305至15466762区段的单倍重复,大小约1 M。将正常对照样本与先证者及父母样本进行同组qPCR检测,以ALB基因为内参基因,对目标PMP22基因1至5外显子的拷贝数进行检测(荧光定量PCR法)显示,患儿及患儿母亲PMP22基因1至5外显子的拷贝数与正常对照的比值约为1.5,提示患儿及患儿母亲PMP22基因的1至5外显子存在单倍重复;患儿父亲PMP22基因1至5外显子的拷贝数与正常对照的比值约为1.0,提示患儿父亲PMP22基因的1至5外显子拷贝数正常(图1)。结合患儿的临床表现、EMG及基因报告结果,临床诊断为CMT。给予甲钴胺片口服营养神经,并转至当地医院行相关康复训练。随访1年余,患儿步态异常及弓形足症状有好转。
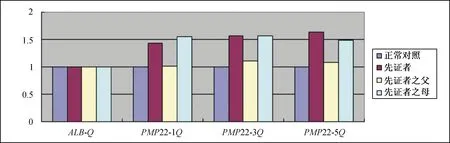
图1 PMP22基因外显子1至5单倍重复qPCR验证结果。ALB-Q:内参基因;PMP22-1Q:PMP22基因外显子1;PMP22-3Q:PMP22基因外显子3;PMP22-5Q:PMP22基因外显子5
2 讨论CMT也称为遗传性运动感觉神经病(HMSN),是一组以进行性肌无力、感觉异常和神经传导功能异常为特征的遗传性神经病,根据正中神经或尺神经的运动神经传导速度(MNCV)可分为CMT1(脱髓鞘型)(MNCV<38 m/s)及CMT2(轴突型)(MNCV≥38 m/s)。但神经传导速度降低程度不能反映疾病的临床严重程度[3-4]。既往报道CMT患病率为1/2 500,挪威阿克尔修斯县东部的CMT患病率是1/1 214,是挪威西部的发病率(1/2 500)的2倍[5]。西班牙、意大利、冰岛和日本的发病率分别为1/3 500、1/5 700、1/8 300和1/9 200[5]。最常见的遗传模式是常染色体显性遗传,也有X连锁遗传和常染色体隐性遗传,最常涉及的基因是PMP22(>50%)。由于没有严格的基因型-表型相关性,CMT的临床表现具有异质性。典型的临床表现为肢体远端无力、感觉、腱反射减弱或消失以及足部畸形,双膝关节以下明显肌萎缩,呈“仙鹤腿”。患者常在20岁之前发病,但疾病临床进展缓慢,一般不会影响预期寿命[2,6]。有些患者可出现脊柱侧凸、感觉和自主神经受累、呼吸损害、上肢受累、视觉或听力障碍、锥体束征和智力迟缓等临床表现,这些临床表现可用于确定某些特定亚型[4]。目前,仅体格检查和电生理检查可用于评估CMT患者的疾病严重程度。
本例患儿腰椎轻度侧弯,S1椎体可见隐裂;EMG示上下肢多发运动、感觉周围神经损害(脱髓鞘合并轴索损害);患儿母亲、外婆均有不同程度弓形足及步态异常。检查结果及家族史高度提示患儿为遗传性周围神经病,进一步行全外显子组测序显示,患儿17号染色体存在大片段CNV,是位于chr17:14095305至15466762区段的单倍重复,大小约为1 Mb。这不仅与常见的1.5 Mb的染色体重复有所不同,且尚未见报道。荧光定量PCR验证显示,患儿及患儿母亲PMP22基因的1至5外显子存在单倍重复,患儿父亲PMP22基因的1至5外显子拷贝数正常,符合常染色体显性遗传致病机制。理论上,该家庭中母亲生育任一胎,均有50%的患病风险。既往PMP22基因重复突变多采用多重连接探针扩增方法进行检测,但该方法不能检测出未知的点突变。本例采用全外显子测序技术结合实时荧光定量PCR检测验证,可以为未知的点突变患儿提供精准分子诊断,有效避免了漏诊、错诊。
目前尚无针对CMT的有效药物,临床仅采用对症治疗。目前,无论是运动和矫形器治疗,还是神经节苷脂、肌酸以及口服抗坏血酸的药理学方法,都没有显示出对CMT的明确效果[7]。由于本病临床进展缓慢,且缺乏有效评估指标,导致无法准确评估口服抗坏血酸的疗效[8]。对有家族史的妊娠期妇女进行遗传咨询及产前诊断是减少CMT发病率的有效手段,通过早期干预,尽快、尽早的康复训练可积极改善预后,避免严重临床后遗症的产生。
相信随着对CMT发病机制与致病基因的进一步研究,对评估指标进一步完善,对疾病进行早期诊断与早期干预,CMT的治疗前景会越来越开阔,治愈CMT将会成为一种可能。
