选择性烷基化合成5-取代的淫羊藿素衍生物
2021-06-28崔晗琦赵长阔董维维赵缘财王先恒
崔晗琦,赵长阔 ,董维维,赵缘财,王先恒
(1.遵义医科大学 基础药理教育部重点实验室暨特色民族药教育部国际合作联合实验室,贵州 遵义 563099;2.遵义医科大学 药学院,贵州 遵义 563099)
淫羊藿是一种传统的中草药,《本草纲目》中称其有“益精气,坚筋骨,补腰膝,强心力”之功效[1]。近期有关研究表明,淫羊藿中含有的主要活性成分,如淫羊藿素、淫羊藿苷、和淫羊藿次苷Ⅱ,发现它们的抗肿瘤效果均较为显著,有文献报道该类衍生物对不同类型的肿瘤,例如前列腺癌[2]、宫颈癌[3]、胃癌[4]、肝癌[5]、肺癌[6]及骨肉瘤等都具有不同程度的抑制作用[7-9]。通过淫羊藿苷(Icariin)的代谢研究表明,其口服吸收率低,生物利用度只有12%,在体内经过纤维素酶代谢将苷元结构水解得到淫羊藿素(Icaritin)并发挥作用;将淫羊藿苷大鼠静脉给药后,在小肠及尿中发现其代谢产物有淫羊藿次苷Ⅱ(IcarisideⅡ)和淫羊藿素(Icaritin)[10]。淫羊藿素,化学名称为3,5,7-三羟基-2-(4-甲氧基苯基)-8-(3-甲基丁-2-烯基)-4H-色烯-4-酮(见图1)。
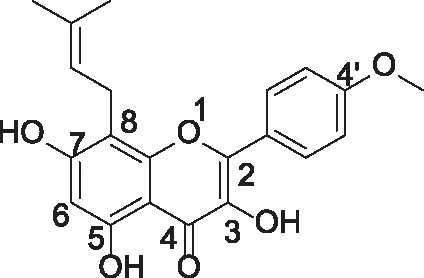
图1 Icaritin的结构式
淫羊藿素具有许多药理和生物活性,如神经保护作用[11],成骨分化促进和破骨细胞分化抑制活性[12],人类前列腺的生长抑制癌PC-3细胞[13],诱导人肝癌HepG2细胞凋亡[14]及多药耐药逆转活性[15]。尤其是,淫羊藿素表现出特殊的雌激素活性,在微摩尔浓度下刺激ER阳性乳腺癌MCF-7细胞的增殖[16];而在微摩尔浓度范围内,淫羊藿素抑制ER阳性(MCF-7)和ER阴性(MDA-MB-453)乳腺癌细胞的生长[17]。这些特征使淫羊藿素成为SERMs双向调节的抗乳腺癌药物。
与许多其他具有生物活性的黄酮类化合物一样,淫羊藿素的主要缺点是其在水中的溶解性差,这导致其在体内较低的生物利用度,其在给药后主要以结合形式存在,通过粪便排泄[18]。这很大的限制了它在临床方面上的应用,因此就要对其结构进行修饰,得到相应的衍生物,从而提高生物利用度,并且提高其抗生物活性。
本次结构修饰目的是找到合适的条件,对淫羊藿素的3,5,7位上的三个羟基进行选择性烷基化,对其结构相应的位置上的基团进行修饰,并不断地改变烷基化试剂,并可以从中找到较有活性的淫羊藿素的衍生物,提高生物利用度并得到更好的抗肿瘤效果。
1 材料与方法
1.1 材料和仪器 淫羊藿素(>98% ,上海融禾医药科技发展有限公司);溴丙烯(分析纯,北京偶合科技有限公司);溴丙炔(分析纯,武汉长成化成科技发展有限公司);碘甲烷(分析纯,南京皇泰医药科技有限公司);溴乙烷(分析纯,上海嵩爰化工科技有限公司);溴代正丁烷、1,4-二溴丁烷(分析纯,上海易恩化学技术有限公司);1-溴戊烷(分析纯,上海迈瑞尔化学技术有限公司);1-碘丙烷(分析纯,上海麦克林生化科技有限公司);氯化苄(分析纯,石家庄苏兴生物科技有限公司);石油醚(分析纯,天津市富宇精细化工有限公司);乙酸乙酯(分析纯,江苏强盛功能化学有限公司);丙酮(分析纯,川东化工);柱层析用硅胶(粒度 200 ~ 300 目,青岛海洋化工厂)、硅胶薄层板(青岛海洋化工厂)。
仪器采用 81-2型暗箱三用紫外分析仪(上海司乐仪器有限公司);Varian 400MHz 核磁共振波谱仪(TMS 作内标,安捷伦科技有限公司)。
1.2 方法
1.2.1 合成路线(见图2)
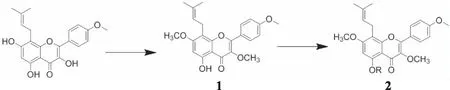
图2 5-取代淫羊藿素衍生物的合成路线
1.2.2 5-羟基-3,7-二甲氧基-2-(4-甲氧基苯基)-8-(3-甲基丁-2-烯-1-基)-4H-色烯-4-酮的合成 将淫羊藿素(50 mg,0.14 mmol)装入到25 ml圆底烧瓶中,再向其加入K2CO3(39.28 mg,0.284 mmol),加入丙酮溶剂(3 mL),最后滴加碘甲烷(0.010 ml,0.168 mmol),于60℃下加热回流反应,一开始固体溶解,溶液会变澄清,随着反应会有黄色固体析出,TLC点板跟踪直至反应完全,加入水和乙酸乙酯进行萃取,合并有机相,用无水硫酸镁吸收水分,真空减压浓缩,将旋出物经过柱层析(PE:EA=6∶1)得到黄色固体40 mg,产率为80%。合成路线(见图3)。
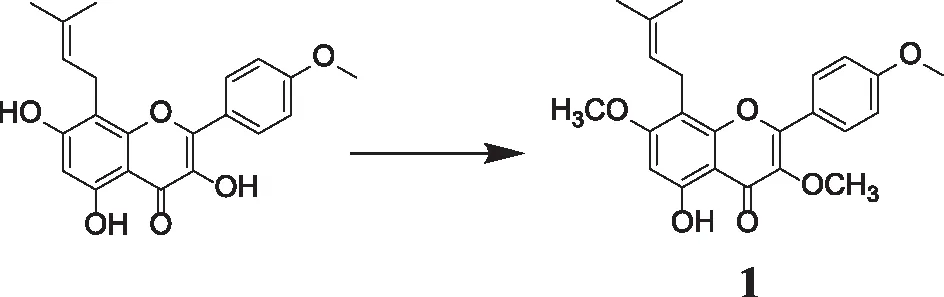
图3 淫羊藿素3,7-OH的甲基化反应
1.2.3 化合物2a5-(烯丙氧基)-3,7-二甲氧基-2-(4-甲氧基苯基)-8-(3-甲基丁-2-烯-1-基)-4H-色烯-4-酮的合成 向25 mL封管中,依次投入化合物1(50 mg,0.13 mmol)、K2CO3(35.93 mg,0.26 mmol)、丙酮溶剂(3 mL),常温搅拌10 min,缓慢滴加卤代烃试剂溴丙烯(0.013 mL,0.156 mmol)。滴毕,升温至65 ℃并保温反应6 h。反应结束后,旋干丙酮,随后加入水和乙酸乙酯萃取3次,合并有机相,真空减压浓缩,将旋出物经过柱层析(PE:EA=3∶1)得到白色固体2a20 mg,产率为40%。按化合物2a类似的方法分别制备目标化合物2b~2h(图4,数据见表1,表2)。
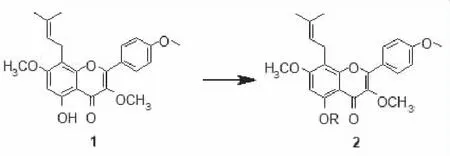
图4 5-取代淫羊藿素的合成
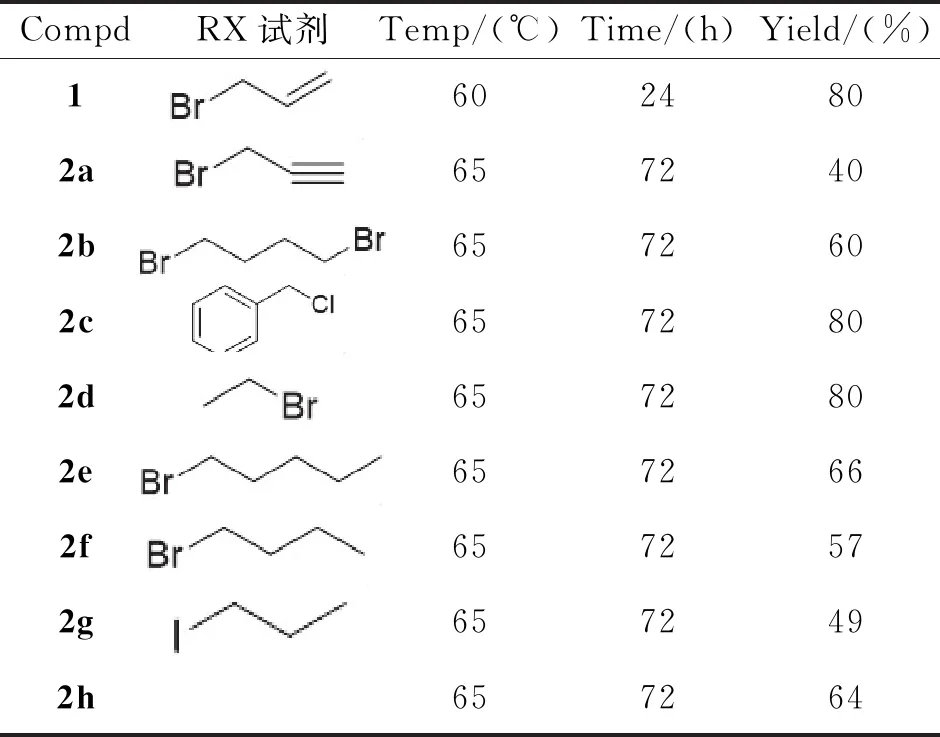
表1 反应条件的优化
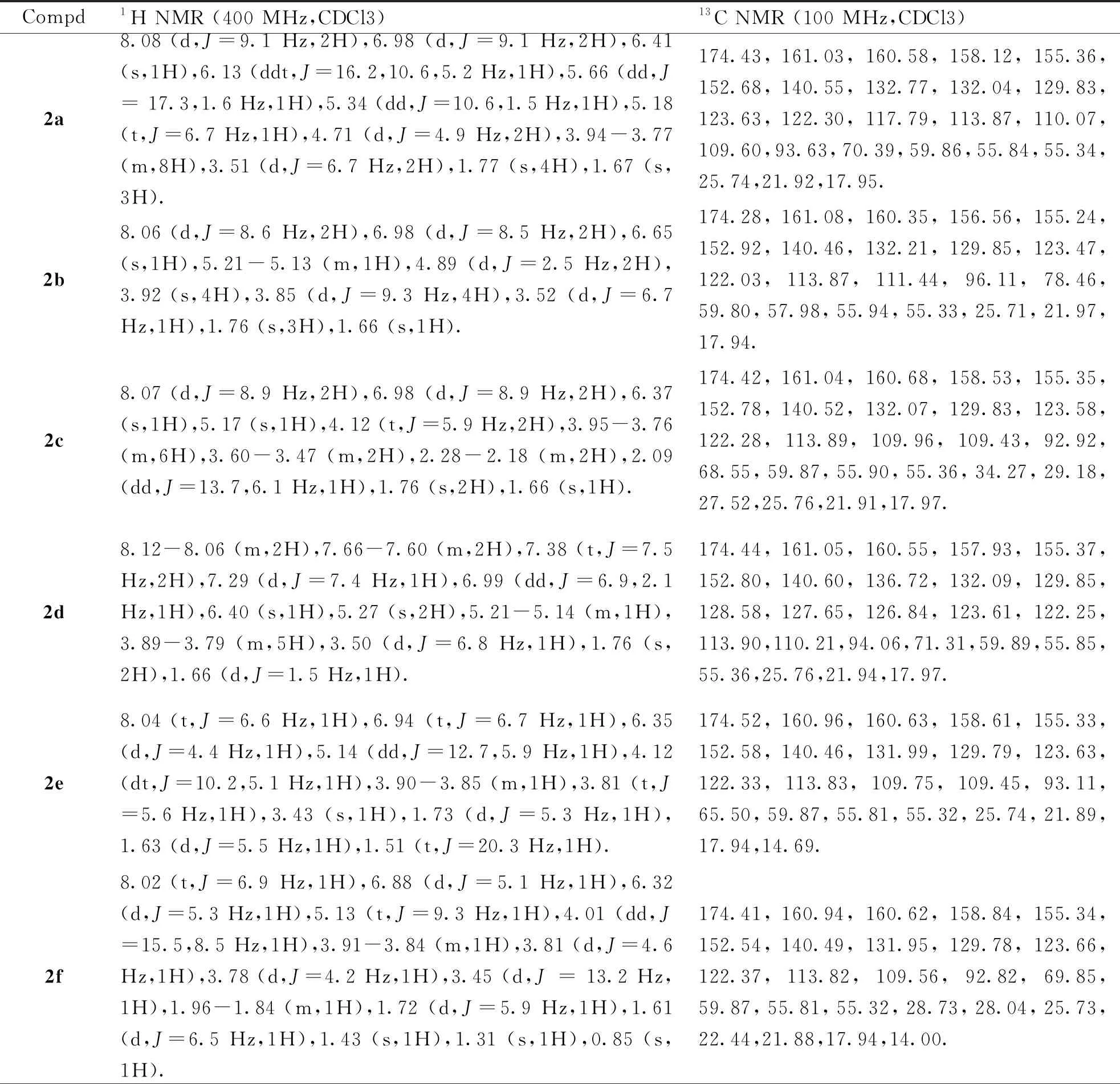
表2 化合物2a-2h的核磁共振光谱数据
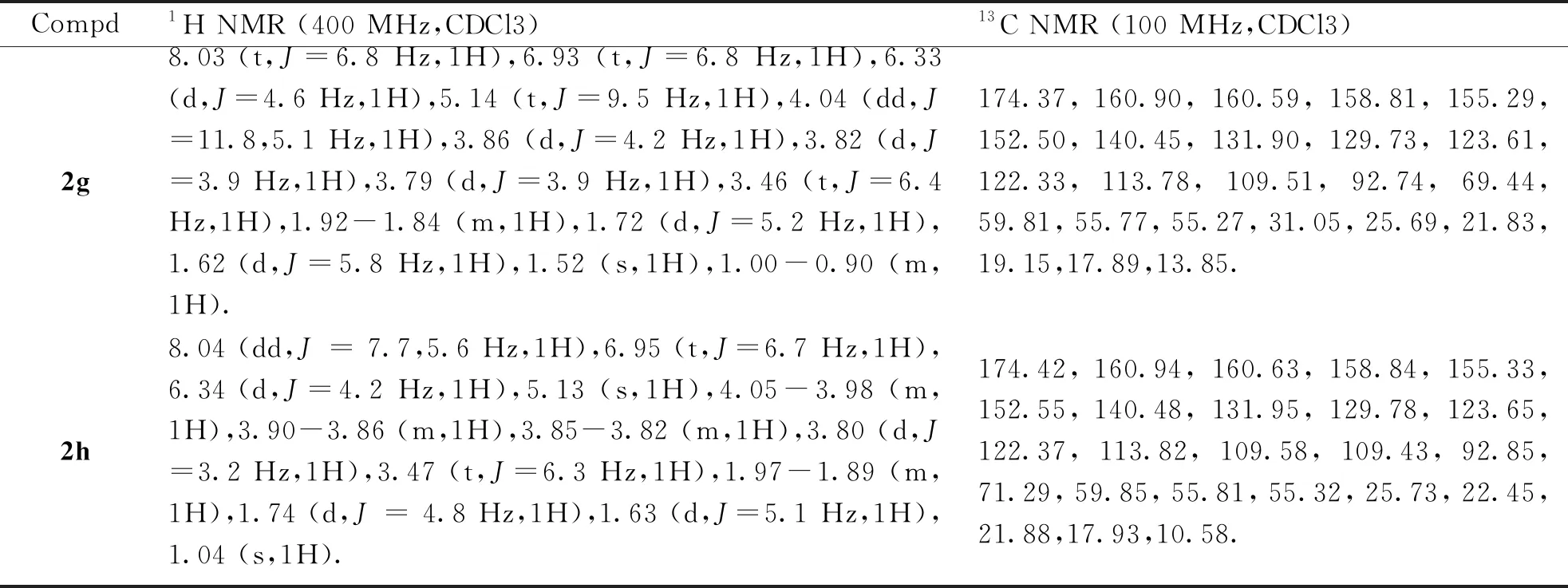
续表
2 结果
在第一步反应中,淫羊藿素与碘甲烷的烷基化试剂得到双位取代产物的反应产率较高;第二步反应中,采取高温高压反应条件,合成目标化合物的时间较快;可以通过与不同的烷基化试剂反应,不同的反应条件,对淫羊藿素的C-3,C-5,C-7位上的3个羟基进行选择性烷基化,以期找到较有活性的淫羊藿素的衍生物,提高生物利用度并得到更好的药效。
3 讨论
3.1 5-羟基-3,7-二甲氧基-2-(4-甲氧基苯基)-8-(3-甲基丁-2-烯-1-基)-4H-色烯-4-酮的合成 根据文献的查阅,所采取的烷基化试剂是硫酸二甲酯,得到C-3的被甲氧基取代的淫羊藿素衍生物。实验过程中,发现除此之外还会伴随有C-3,C-7都被甲氧基取代的淫羊藿素衍生物出现,两个产物难以通过硅胶柱分离,且所得产率不高。反应尝试将烷基化试剂替换为碘甲烷,直接得到C-3,C-7都进行甲氧基取代的双位取代烷基化中间产物,产率高达80%。由于碘离子体积大,容易极化,对于甲基化反应碘甲烷活性要高于硫酸二甲酯;本研究中采用碘甲烷与淫羊藿素进行C-3,C-7两个羟基的甲基化保护后,再与相应卤代烃进行C-5的5-OH进行烷基化取代。
3.2 化合物2a-2h合成 把化合物1作为原料在碱性条件下与不同烷基化试剂进行反应,试图得到C-3,C-7被甲氧基取代,C-5被不同的烷基取代的淫羊藿衍生物。在该步反应中,C-4上的羰基与C-5羟基之间形成稳定的分子内氢键,因此C-5的羟基比C-3,C-7上的羟基活性更差,所以采取了高温高压的条件。
