美沙西汀-甲氧基-13C及其类似物的合成
2021-10-23代德胜王春玲魏翠雯沈桂富
陈 凯,代德胜,王春玲,魏翠雯,卿 晶,沈桂富
(深圳市中核海得威生物科技有限公司,广东 深圳 518057)
呼气试验是指呼气成分直接测定或测定摄入特定化合物后呼气中的标志气体,实现对机体生理、病理状态的非侵入性判断。呼气试验技术简单、方便,受检者依从性好、容易操作,可完成一般方法难以进行的诊断[1]。目前已经应用的临床技术包括一氧化氮呼气检测哮喘[2]、13/14C-尿素呼气试验诊断幽门螺杆菌感染[3]、呼气试验测定红细胞寿命、13C-螺旋藻诊断胃排空功能障碍[4]等。其中13/14C-尿素呼气试验已经成为幽门螺杆菌感染诊断应用最广泛的方案,产业规模在中国已超过20亿每年。
肝脏是人体最重要的代谢器官之一,担负着解毒、营养物质代谢、蛋白质合成、胆汁分泌等重要的生化功能,肝脏功能的准确性测定有重要的临床意义。目前常用的检测方法包括血液生化检查、药物动力学实验和代谢实验检测等。鉴于13/14C-尿素呼气试验诊断幽门螺杆菌准确度高、市场推广好,13C-标记化合物呼气试验检测肝部功能成为一个新的研究热点。例如:13C-氨基比林[5]、L-[1-13C]-苯丙氨酸[6]、咖啡因[3-甲基-13C][7]、美沙西汀-甲氧基-13C[8-9]、D-半乳糖-1-13C[10]等,其中美沙西汀-甲氧基-13C较其他的标记化合物具有用量小、几乎无毒、代谢快、适用性广等优点,是研究最活跃的肝功能呼气试验底物[11-12]。
鉴于美沙西汀-甲氧基-13C呼气试验的特点,近些年关于美沙西汀-甲氧基-13C评估肝功能、诊断肝硬化和肝移植患者术前与术后的功能评价[13]等方面的呼气诊断临床研究很多,多项实验处于临床Ⅱ期和临床Ⅲ期。目前一般以碘甲烷-13C为标记原料,收率高,是一种常用的合成方法。由于碘甲烷-13C毒性大,运输、储存管理要求非常严格,且每摩尔碘甲烷-13C的价格远高于甲醇-13C,通过甲醇-13C合成美沙西汀-甲氧基-13C得到更多关注[14-15]。
对甲氧基苯甲酸-甲氧基-13C是美沙西汀-甲氧基-13C的类似物,可作为药物、防腐剂以及香料等稳定同位素内标试剂的重要中间体。对甲氧基苯甲酸常见的合成方法以硫酸二甲酯为甲基化试剂,该合成方案无法直接应用于对甲氧基苯甲酸-甲氧基-13C的合成。2018年,上海稳定同位素工程技术研究中心的王伟[16]以尼铂金乙酯、碘甲烷-13C为原料,通过酚羟基甲基化、水解乙酯两步反应得到对甲氧基苯甲酸-甲氧基-13C,其中碘甲烷-13C的利用率为79.32%。
本文以Mitsunobu反应作为关键反应,配合简单的官能团转化,以甲醇-13C为标记试剂,合成美沙西汀-甲氧基-13C和对甲氧基苯甲酸-甲氧基-13C,拟为呼气检测肝功能研究的开展提供参考。
1 主要实验材料
1.1 主要试剂
氧羰酰-4-羟基苯胺(纯度>98%)、三苯基膦(纯度>98%)、偶氮二甲酸二乙酯(纯度>98%)、偶氮二甲酸二异丙酯(纯度>98%)、三氟乙酸(纯度>99%)、三乙胺(纯度>99.5%)、乙酰氯(纯度>98%):萨恩化学技术(上海)有限公司;尼泊金甲酯(纯度>98%):上海迈瑞尔化学技术有限公司;甲醇-13C(纯度>98%,同位素丰度为99%13C)、美沙西汀-甲氧基-13C(纯度98.7%,同位素丰度为99%13C,对照品):剑桥同位素实验室;对甲氧基苯甲酸-甲氧基-13C(纯度98%,同位素丰度为99%13C,对照品):武汉纽瑞德特种气体有限公司;乙腈(色谱纯):默克股份两合公司;乙酸乙酯(EA,分析纯)、正己烷(HEX,分析纯)、乙醇(分析纯)、碳酸钠(分析纯)、氯化钠(分析纯)、氢氧化钠(分析纯)、无水硫酸钠(分析纯):国药集团化学试剂有限公司;柱层析硅胶:精制型,200~300目,青岛海洋化工有限公司;除特别说明外,所有试剂均直接使用。
1.2 主要仪器
XPE303S型电子天平:梅特勒-托利多;MS7-H550-Pro数显加热磁力搅拌器恒温磁力搅拌器:大龙兴创实验仪器(北京)股份公司;EYELA东京理化旋转蒸发仪 N-1300系列:东京理化器械株式会社;SHK-Ⅲ循环水式多用真空泵:郑州科泰实验设备有限公司;HB-20耐腐蚀隔膜真空泵:上海沪析实业有限公司;Agilent 1260高效液相色谱仪:安捷伦科技有限公司;核磁共振氢谱(布鲁克400M核磁AVANCE-Ⅲ)检测和高分辨质谱(赛默飞高分辨质谱Q Exactive Focus)检测由北京大学深圳研究生院化学生物学和生物技术学院协助完成。
2 实验方法
2.1 反应机理
Mitsunobu 反应[17]是一种涉及醇羟基活化的亲核取代反应,其反应机理示于图1。醇在偶氮二甲酸二乙酯和三苯基膦的作用下形成活性中间体,该活性中间体被亲核试剂进攻之后碳氧键会发生断裂。依据上述反应原理,甲醇-13C的碳氧键可以被偶氮二甲酸二乙酯、三苯基膦活化,之后酚羟基进攻活化中间体即可完成美沙西汀-甲氧基-13C中醚键的构建。
2.2 路线优化
本研究采用对乙酰氨基酚、甲醇-13C为原料,通过Mitsunobu反应得到美沙西汀-甲氧基-13C(图2)。由于美沙西汀-甲氧基-13C与反应的副产物三苯基氧化膦极性接近,分离困难,不适于进行放大研究。通过优化,将反应原料对乙酰氨基酚更换为极性更小的氧羰酰-4-羟基苯胺。
以氧羰酰-4-羟基苯胺、甲醇-13C为起始原料,首先通过Mitsunobu反应得到化合物3,由于化合物3极性小,很容易与副产物三苯基氧化膦分离。
2.3 美沙西汀-甲氧基-13C合成
美沙西汀-甲氧基-13C的合成路线示于图2。将100 mL乙腈加至250 mL圆底烧瓶中,再依次加入4.18 g氧羰酰-4-羟基苯胺、0.81 mL甲醇-13C和6.28 g三苯基膦,冷却至0 ℃。将4.40 mL偶氮二甲酸二异丙酯缓慢加入反应体系,随后在室温条件下搅拌4 h。体系用30 mL饱和碳酸钠水溶液淬灭,用200 mL乙酸乙酯萃取2次,合并有机相。有机相使用30 mL饱和食盐水清洗,无水硫酸钠干燥,过滤后减压浓缩。剩余物用柱层析色谱(200~300目,洗脱剂为乙酸乙酯∶正己烷=1∶4)纯化,真空干燥后得到白色固体3.12 g。将白色固体溶于60 mL二氯甲烷中,冷却至0 ℃,向反应体系中加入10 mL三氟乙酸。室温搅拌3 h,减压蒸馏除去体系中的溶剂和三氟乙酸。随后将浓缩物溶于80 mL二氯甲烷中,冷却至0 ℃,向反应体系中依次加入4.16 mL三乙胺、1.78 mL乙酰氯,室温反应2 h。使用10 mL饱和氯化铵淬灭反应;反应液用乙酸乙酯100 mL萃取3次,合并有机相,用饱和食盐水20 mL洗涤,无水硫酸钠干燥;过滤、旋干溶剂后用乙醇重结晶,得到固体2.10 g,即为美沙西汀-甲氧基-13C。

图2 美沙西汀-甲氧基-13C的合成路线Fig.2 Synthetic route of of methacetin-methoxy-13C
2.4 对甲氧基苯甲酸-甲氧基-13C合成
对甲氧基苯甲酸-甲氧基-13C合成路线示于图3。将100 mL乙腈加至250 mL圆底烧瓶中,再依次加入3.04 g尼泊金甲酯、0.81 mL甲醇-13C和6.28 g三苯基膦,冷却至0 ℃。将4.40 mL偶氮二甲酸二异丙酯缓慢加入反应体系,随后在室温条件下搅拌4 h。体系用30 mL饱和碳酸钠水溶液淬灭,用200 mL乙酸乙酯萃取2次,合并有机相。有机相使用30 mL饱和食盐水清洗,无水硫酸钠干燥,过滤后减压浓缩。剩余物用柱层析色谱(200~300目,洗脱剂为乙酸乙酯∶正己烷=1∶8)纯化,真空干燥后得到白色固体2.65 g。白色固体溶于80 mL乙腈/水中,冷却至0 ℃,加入1.60 g氢氧化钠。室温反应12 h,冷却至0 ℃,使用6 mol/L的盐酸酸化至体系pH为1。使用200 mL乙酸乙酯萃取反应液3次,有机相使用30 mL饱和食盐水清洗,无水硫酸钠干燥,过滤后减压浓缩。浓缩后的固体用80 ℃的乙醇溶解,冷却至2~8 ℃结晶。过滤干燥后得到产品2.26 g,即为对甲氧基苯甲酸-甲氧基-13C。

图3 对甲氧基苯甲酸-13C的合成路线Fig.3 Synthetic route of 4-methoxybenzoic acid-methoxy-13C
3 结果与讨论
3.1 条件优化
溶剂和活化试剂对反应产率的影响结果列于表1,由表1可以看出,乙腈作为反应溶剂时收率高于用四氢呋喃和二氯甲烷,偶氮二甲酸二异丙酯(DIAD)的反应效果好于偶氮二甲酸二乙酯(DEAD)。表1中各实例中反应液浓度均为0.25 mmol/mL,反应时间均为4 h。使用三氟乙酸脱除叔丁氧羰基,最后在碱性条件下对氨基乙酰化即可得到美沙西汀-甲氧基-13C。该合成方案可放大,操作简单,可作为合成美沙西汀-甲氧基-13C工艺化的方案。
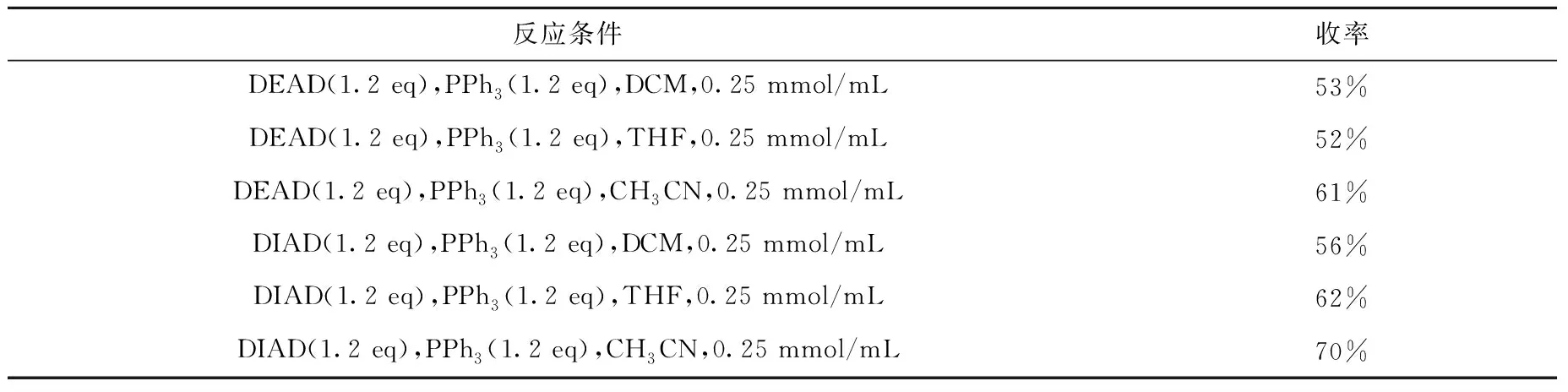
表1 Mitsunobu反应条件优化Table 1 Optimization of Mitsunobu reaction conditions
在美沙西汀-甲氧基-13C合成研究的基础上,以尼泊金甲酯、甲醇-13C为原料,通过Mitsunobu反应、水解甲酯两步反应得到对甲氧基苯甲酸-甲氧基-13C。相比以碘甲烷-13C作为标记原料的合成方法,本方法降低了成本。
3.2 美沙西汀-甲氧基-13C的结构表征和含量测定
美沙西汀-甲氧基-13C合成样品的核磁共振氢谱示于图4。1H NMR(400 MHz, Acetone-d6)δ9.02(s,1H),7.60-7.45(m,2H),6.92-6.75(m,2H),3.92(s,2H),3.56(s,2H),2.03(s,3H)。美沙西汀-甲氧基-13C的高分辨质谱数据为ESI-MS:[M+H]+=167.089 6,[M+Na]+=189.071 6,结构表征数据与对照品一致。

图4 美沙西汀-甲氧基-13C的核磁共振氢谱Fig.4 1H NMR of methacetin-methoxy-13C
在合成样品纯度测定方面,本文以纯度为98.7%的美沙西汀-甲氧基-13C样品作为对照品。通过分析对照品和合成样品的高效液相数据(表2),根据外标法计算出合成样品的纯度为99.3%。经高分辨质谱检测美沙西汀-甲氧基-13C合成样品的同位素丰度为99.2%13C,符合标记化合物丰度和纯度的要求。
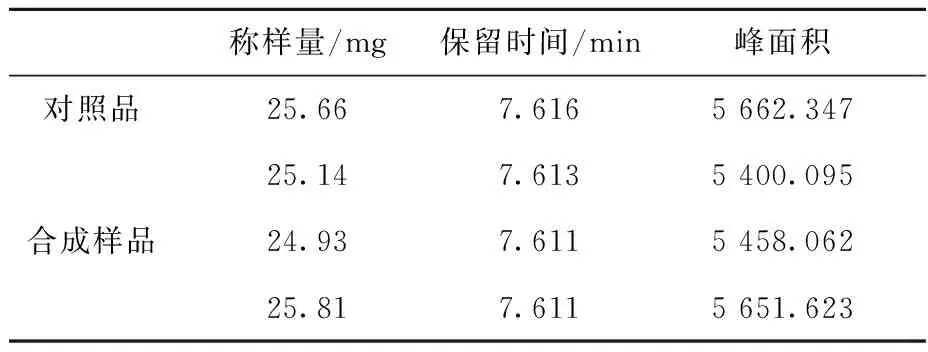
表2 美沙西汀-甲氧基-13C的高效液相色谱分析Table 2 HPLC data of methacetin-methoxy-13C
3.3 对甲氧基苯甲酸-甲氧基-13C的结构表征和含量测定
对甲氧基苯甲酸-甲氧基-13C合成样品的核磁共振氢谱示于图5。1H NMR(400 MHz, Acetone-d6)δ8.04-7.92(m, 2H),7.07-6.92(m,2H),4.06(s,1.5H),3.70(s,1.5H)。美沙西汀-甲氧基-13C的高分辨质谱数据为ESI-MS:[M-H]+=152.042 2(负离子质谱),结构表征数据与对照品一致。
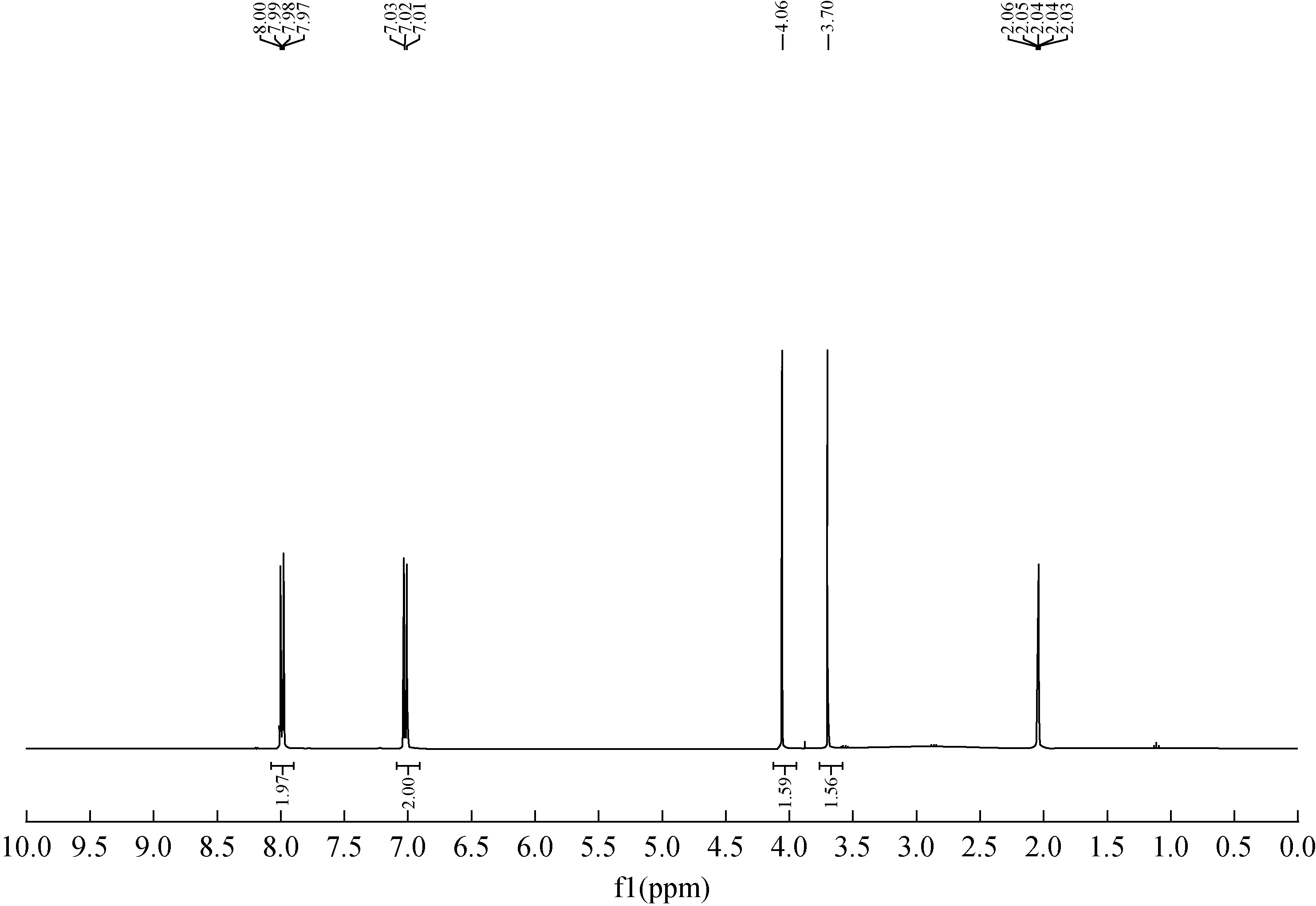
图5 对甲氧基苯甲酸-甲氧基-13C的核磁共振氢谱Fig.5 1H NMR of 4-methoxybenzoic acid-methoxy-13C
在产物纯度测定方面,本文以纯度为98%的对甲氧基苯甲酸-甲氧基-13C样品作为对照品。通过对照品和合成样品的高效液相色谱分析(表3),根据外标法计算出对甲氧基苯甲酸-甲氧基-13C合成样品的纯度为101.0%,符合纯度要求。

表3 对甲氧基苯甲酸-甲氧基-13C的高效液相色谱分析Table 3 HPLC data of 4-methoxybenzoic acid-methoxy-13C
4 结论
本研究通过Mitsunobu反应、官能团转化合成了美沙西汀-甲氧基-13C及其类似物对甲氧基苯甲酸-甲氧基-13C。美沙西汀-甲氧基-13C合成样品的纯度为99.2%,同位素丰度为99.2%13C,以甲醇-13C计算收率为63%。对甲氧基苯甲酸-甲氧基-13C合成样品的纯度为101.0%,同位素丰度为99.2%13C,以甲醇-13C计算收率为67%。美沙西汀-甲氧基-13C和对甲氧基苯甲酸-甲氧基-13C的同位素丰度相对于标记原料甲醇-13C未发生明显降低。美沙西汀-甲氧基-13C的合成为进一步的呼气试验研究提供了基础,同时该方法还可应用于其他芳基醚类标记化合物如非那西汀-乙氧基-1-13C等。
